


Revisar a literatura que aborda a relação entre os parâmetros de crescimento e nutricionais com a função pulmonar em pacientes pediátricos com fibrose cística.
Fontes de dadosDados foram coletados de artigos publicados nos últimos 15 anos em Inglês,Português e Espanhol através de pesquisa nas bases de dados eletrônicas – PubMed, Cochrane, Medline, Lilacs e Scielo – usando as palavras‐chave: fibrose cística, crescimento, nutrição, função pulmonar utilizando combinações variadas. Os artigos que analisaram a associação de longo prazo entre parâmetros de crescimento e nutricionais, com ênfase em crescimento, com doença pulmonar em fibrose cística, foram incluídos, sendo excluídos aqueles que analisaram apenas a relação entre os parâmetros nutricionais e fibrose cística e aqueles em que o objetivo era descrever a doença.
Síntese dos dadosSete estudos foram incluídos, com um total de 12.455 pacientes. Seis relataram relação entre parâmetros de crescimento e função pulmonar, incluindo um estudo que analisou apenas a associação de parâmetros de crescimento com a função pulmonar, e todos os sete relataram associação entre parâmetros nutricionais e função pulmonar.
ConclusõesA revisão sugere que a gravidade da doença pulmonar, determinada por espirometria, está associada com crescimento corporal e o estado nutricional em fibrose cística. Assim, a intervenção nesses parâmetros pode contribuir para um melhor prognóstico e expectativa de vida em pacientes com fibrose cística.
To review the literature addressing the relationship of growth and nutritional parameters with pulmonary function in pediatric patients with cystic fibrosis.
Data sourceA collection of articles published in the last 15 years in English, Portuguese and Spanish was made by research in electronic databases – PubMed, Cochrane, Medline, Lilacs and Scielo – using the keywords cystic fibrosis, growth, nutrition, pulmonary function in varied combinations. Articles that addressed the long term association of growth and nutritional parameters, with an emphasis on growth, with pulmonary disease in cystic fibrosis, were included, and we excluded those that addressing only the relationship between nutritional parameters and cystic fibrosis and those in which the aim was to describe the disease.
Data synthesisSeven studies were included, with a total of 12,455 patients. Six studies reported relationship between growth parameters and lung function, including one study addressing the association of growth parameters, solely, with lung function, and all the seven studies reported relationship between nutritional parameters and lung function.
ConclusionsThe review suggests that the severity of the lung disease, determined by spirometry, is associated with body growth and nutritional status in cystic fibrosis. Thus, the intervention in these parameters can lead to the better prognosis and life expectancy for cystic fibrosis patients.
A fibrose cística (FC) é a doença genética letal mais comum nas populações brancas. Ela é causada por uma mutação em um gene que codifica a proteína cystic fibrosis transmembrane conductance regulator (CFTR), que é expressa em muitas células epiteliais e do sangue e funciona principalmente como um canal de cloreto.1 A doença pulmonar é a manifestação mais importante na FC e o principal fator na morbidade e mortalidade da doença. A resposta na doença pulmonar é mediada pela CFTR anormal,2 genes modificadores,3–8 infecções das vias aéreas e inflamação,9 provavelmente afeta o peso e a altura, devido à supressão do apetite e ao gasto energético aumentado.
A desnutrição e restrição de crescimento também são frequentes e estão relacionadas com o comprometimento da função pulmonar em um círculo vicioso: pacientes desnutridos tendem a apresentar pior função pulmonar e pacientes com doença pulmonar grave tendem a crescer menos. Embora essas relações já tenham sido relatadas,10,11 há poucas análises de longo prazo em relação ao cumprimento das metas de crescimento e nutrição para o curso da função pulmonar desde a infância até a idade adulta.
Nesse contexto, o objetivo deste estudo foi analisar estudos de longo prazo que comparam parâmetros de crescimento e nutrição (com ênfase em crescimento) com a função pulmonar em pacientes com FC e avaliar a relação entre esses fatores.
MétodoFoi feita uma revisão da literatura dos últimos 15 anos (2000‐2015) sobre a relação entre crescimento e parâmetros nutricionais e função pulmonar. A busca de referências em inglês, espanhol e português foi foi feita por meio de bases de dados eletrônicas – PubMed, Medline, Cochrane, Lilacs e Scielo – com os descritores FC, crescimento, crescimento corporal, função pulmonar, em combinações variadas e em suas traduções correspondentes para inglês e espanhol. Revisões sobre o tema também foram consultadas, assim como listas de referências de todos os artigos, em busca de novos estudos.
Após essa etapa, iniciamos a avaliação dos artigos com a análise de títulos e resumos. O primeiro critério de inclusão foi a identificação de estudos potencialmente relevantes, considerando‐se aqueles que comparavam os parâmetros de crescimento com função pulmonar. Nesse caso, foram excluídos os estudos em que os objetivos eram comparar o peso e/ou ganho de altura, sem relação com a função pulmonar, e aqueles cujo objetivo era descrever apenas a FC.
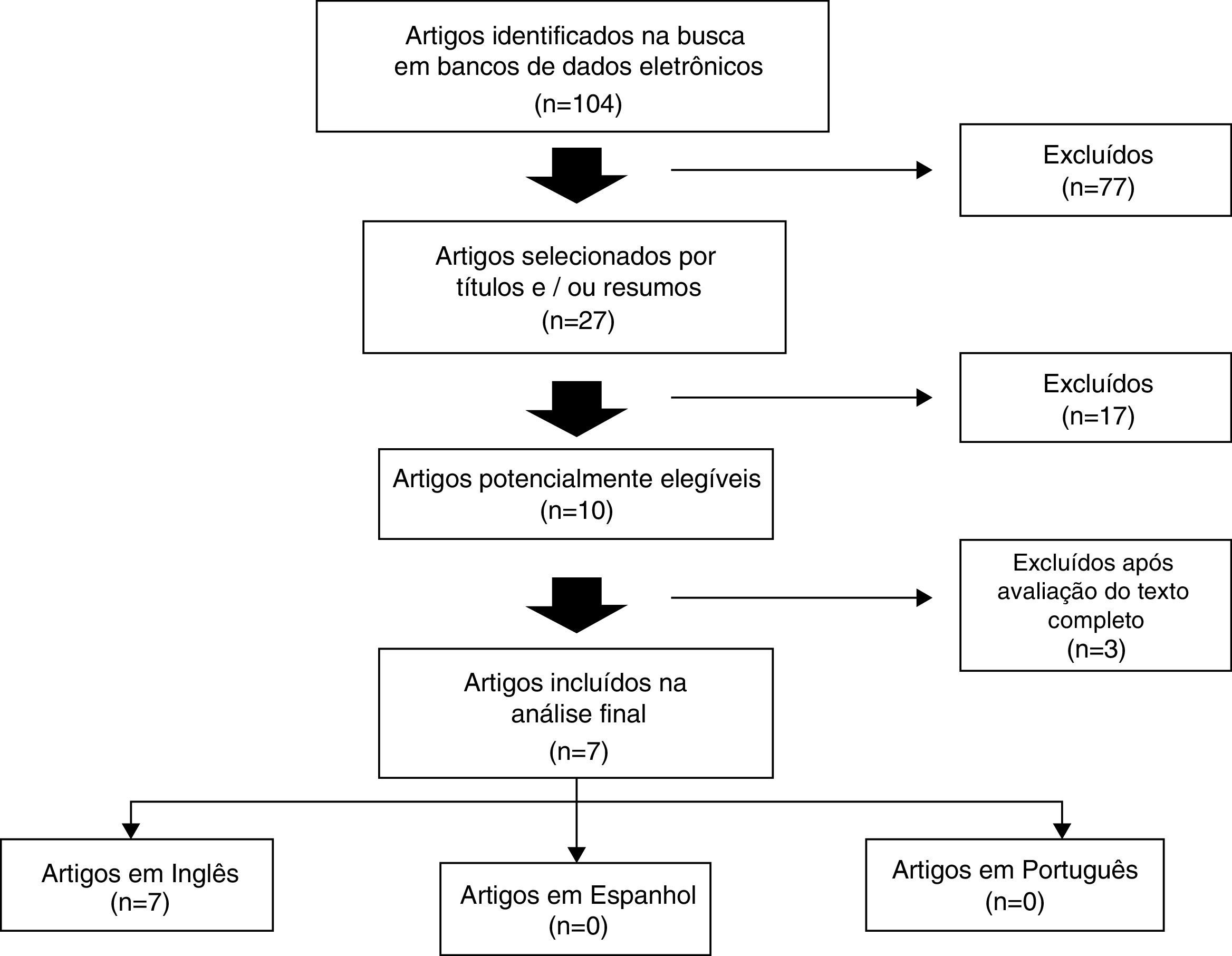
Na primeira busca, 104 artigos foram encontrados. Ao avaliar títulos e resumos, os seguintes critérios de recuperação para artigos completos foram: estudos de coorte, longitudinais, transversais, descritivos e prospectivos, cujos resultados avaliaram a relação entre função pulmonar e os parâmetros de crescimento em pacientes com FC, excluindo aqueles que, apesar de aparecer nos resultados da pesquisa, não abordaram o assunto sob esse ponto de vista. Nessa etapa, 27 artigos foram selecionados. A revisão foi concluída com a leitura dos artigos completos e, no manuscrito final, sete artigos foram incluídos,12–18 todos em inglês (fig. 1; tabelas 1 e 2).

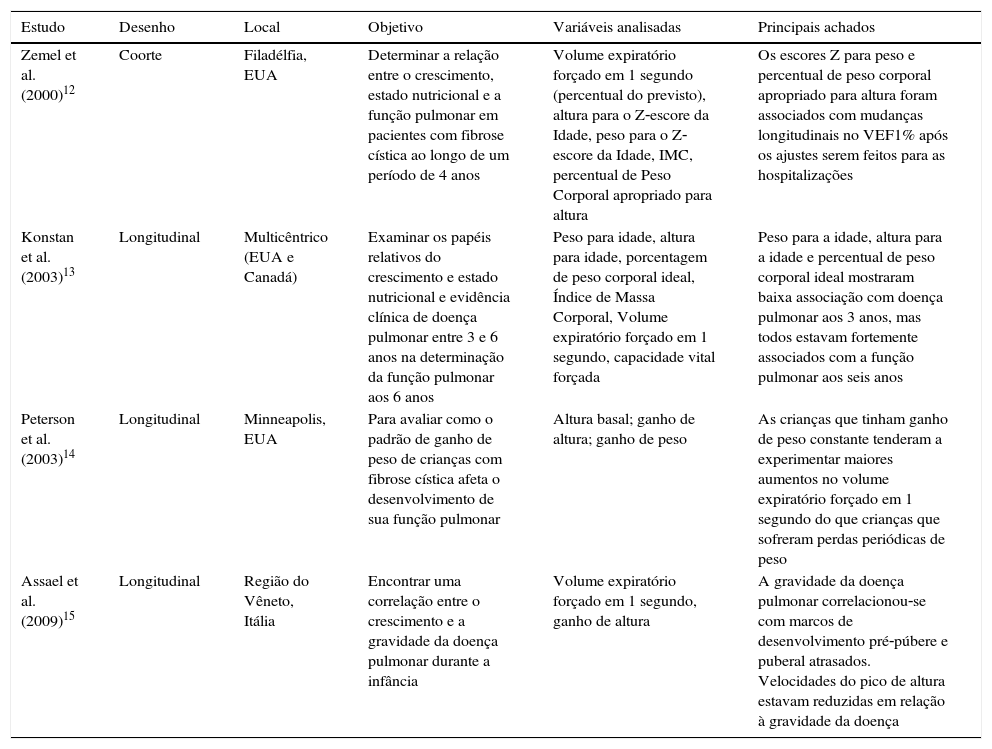
Descrição dos estudos incluídos na revisão sistemática, publicados antes de 2010, distribuídos por autor, desenho, local do estudo, objetivo ou hipótese, as principais variáveis estudadas e principais achados
| Estudo | Desenho | Local | Objetivo | Variáveis analisadas | Principais achados |
|---|---|---|---|---|---|
| Zemel et al. (2000)12 | Coorte | Filadélfia, EUA | Determinar a relação entre o crescimento, estado nutricional e a função pulmonar em pacientes com fibrose cística ao longo de um período de 4 anos | Volume expiratório forçado em 1 segundo (percentual do previsto), altura para o Z‐escore da Idade, peso para o Z‐escore da Idade, IMC, percentual de Peso Corporal apropriado para altura | Os escores Z para peso e percentual de peso corporal apropriado para altura foram associados com mudanças longitudinais no VEF1% após os ajustes serem feitos para as hospitalizações |
| Konstan et al. (2003)13 | Longitudinal | Multicêntrico (EUA e Canadá) | Examinar os papéis relativos do crescimento e estado nutricional e evidência clínica de doença pulmonar entre 3 e 6 anos na determinação da função pulmonar aos 6 anos | Peso para idade, altura para idade, porcentagem de peso corporal ideal, Índice de Massa Corporal, Volume expiratório forçado em 1 segundo, capacidade vital forçada | Peso para a idade, altura para a idade e percentual de peso corporal ideal mostraram baixa associação com doença pulmonar aos 3 anos, mas todos estavam fortemente associados com a função pulmonar aos seis anos |
| Peterson et al. (2003)14 | Longitudinal | Minneapolis, EUA | Para avaliar como o padrão de ganho de peso de crianças com fibrose cística afeta o desenvolvimento de sua função pulmonar | Altura basal; ganho de altura; ganho de peso | As crianças que tinham ganho de peso constante tenderam a experimentar maiores aumentos no volume expiratório forçado em 1 segundo do que crianças que sofreram perdas periódicas de peso |
| Assael et al. (2009)15 | Longitudinal | Região do Vêneto, Itália | Encontrar uma correlação entre o crescimento e a gravidade da doença pulmonar durante a infância | Volume expiratório forçado em 1 segundo, ganho de altura | A gravidade da doença pulmonar correlacionou‐se com marcos de desenvolvimento pré‐púbere e puberal atrasados. Velocidades do pico de altura estavam reduzidas em relação à gravidade da doença |
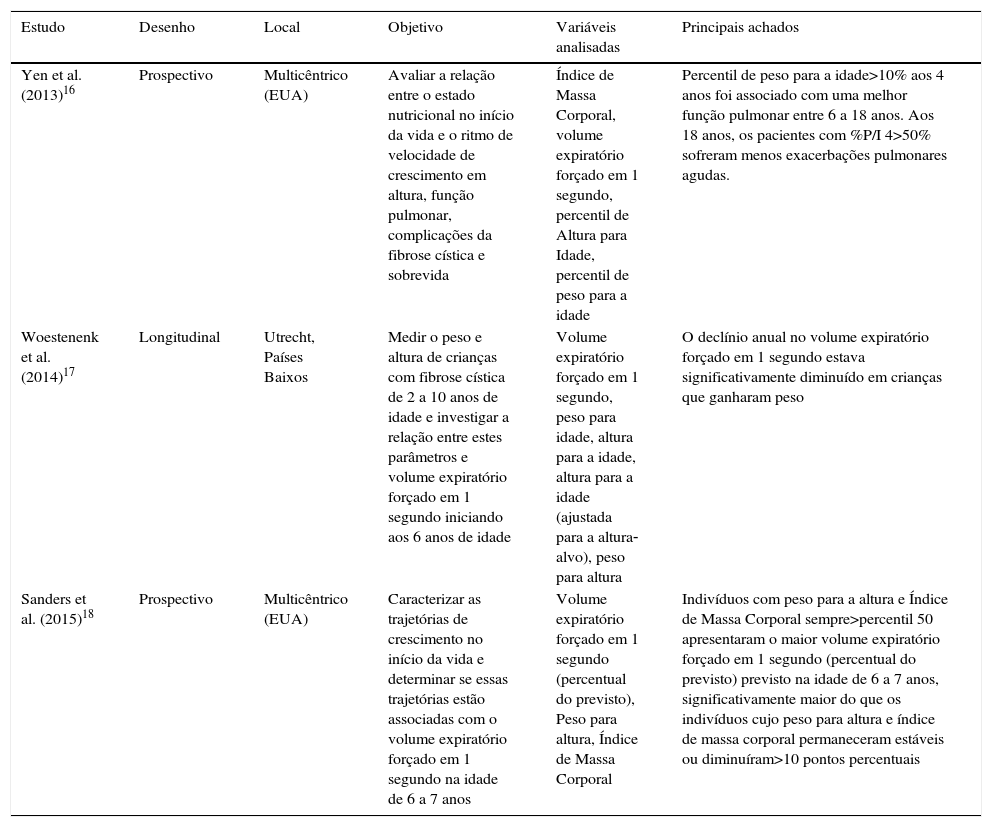
Descrição dos estudos incluídos na revisão sistemática, publicados depois de 2010, distribuídos por autor, desenho, local do estudo, objetivo ou hipótese, principais variáveis estudadas e principais achados
| Estudo | Desenho | Local | Objetivo | Variáveis analisadas | Principais achados |
|---|---|---|---|---|---|
| Yen et al. (2013)16 | Prospectivo | Multicêntrico (EUA) | Avaliar a relação entre o estado nutricional no início da vida e o ritmo de velocidade de crescimento em altura, função pulmonar, complicações da fibrose cística e sobrevida | Índice de Massa Corporal, volume expiratório forçado em 1 segundo, percentil de Altura para Idade, percentil de peso para a idade | Percentil de peso para a idade>10% aos 4 anos foi associado com uma melhor função pulmonar entre 6 a 18 anos. Aos 18 anos, os pacientes com %P/I 4>50% sofreram menos exacerbações pulmonares agudas. |
| Woestenenk et al. (2014)17 | Longitudinal | Utrecht, Países Baixos | Medir o peso e altura de crianças com fibrose cística de 2 a 10 anos de idade e investigar a relação entre estes parâmetros e volume expiratório forçado em 1 segundo iniciando aos 6 anos de idade | Volume expiratório forçado em 1 segundo, peso para idade, altura para a idade, altura para a idade (ajustada para a altura‐alvo), peso para altura | O declínio anual no volume expiratório forçado em 1 segundo estava significativamente diminuído em crianças que ganharam peso |
| Sanders et al. (2015)18 | Prospectivo | Multicêntrico (EUA) | Caracterizar as trajetórias de crescimento no início da vida e determinar se essas trajetórias estão associadas com o volume expiratório forçado em 1 segundo na idade de 6 a 7 anos | Volume expiratório forçado em 1 segundo (percentual do previsto), Peso para altura, Índice de Massa Corporal | Indivíduos com peso para a altura e Índice de Massa Corporal sempre>percentil 50 apresentaram o maior volume expiratório forçado em 1 segundo (percentual do previsto) previsto na idade de 6 a 7 anos, significativamente maior do que os indivíduos cujo peso para altura e índice de massa corporal permaneceram estáveis ou diminuíram>10 pontos percentuais |
Foram avaliados 12.455 pacientes. Considerando o período de início de cada estudo, a faixa etária dos pacientes foi de 0 a 15,3 anos. De acordo com o desenho do estudo, um deles era um estudo de coorte,12 quatro eram estudos longitudinais13–15,17 e dois eram estudos prospectivos.16,18
Os estudos selecionados usaram vários parâmetros de crescimento e nutricionais. Entre os parâmetros nutricionais e de crescimento, o índice de massa corporal (IMC) foi avaliado em cinco estudos,12,14–16,18 apenas a altura foi avaliada em dois estudos,13,15 ganho de altura foi avaliado em dois estudos,13,15 peso para idade (P/I) foi avaliado em dois estudos,14,18 altura para a idade (A/I) foi avaliada em dois estudos.14,16 Os demais parâmetros foram avaliados cada um em um estudo: peso isoladamente,14 porcentagem do peso corporal apropriado para a altura (%PCA),12 porcentagem do peso corporal ideal (PCI%),12 altura para escore‐Z da idade (AZI),12 peso para escore‐Z da idade (PZI),12 altura para a idade ajustada pela altura‐alvo (AI/AA),17 peso para comprimento e os percentis de IMC (PC‐IMC)18 (tabelas 1 e 2).
Todos os estudos usaram volume expiratório forçado em um segundo (VEF1 ou VEF1% previsto) como medida de função pulmonar e apenas um usou a capacidade vital forçada.14
Seis estudos avaliaram a relação entre crescimento e parâmetros nutricionais com a função pulmonar12–14,16–18 e apenas um estudo avaliou a relação de crescimento isolado com a função pulmonar.15
Principais resultadosZemel et al.12 estudaram uma grande amostra (968 crianças) provenientes de vários centros de atendimento nos Estados Unidos por quatro 4 anos. Durante os primeiros três anos de seguimento, os pacientes do sexo feminino mostraram redução em AZI, enquanto os pacientes do sexo masculino aumentaram AZI. Ambos os grupos mostraram diminuição nesse mesmo parâmetro no último ano. Houve diferenças no padrão de mudanças longitudinais no VEF1% e a taxa de declínio foi menor nos pacientes do sexo masculino. Os autores descobriram que AZI era positiva e significativamente associada com VEF1%, bem como PZI e %PCA.
Konstan et al.13 avaliaram, em 931 pacientes, o papel relativo do crescimento e o estado nutricional e a evidência clínica de doença pulmonar desde três a seis anos na determinação da função pulmonar aos seis anos. Os resultados mostraram que significa P/I e A/I em ambas as idades três e seis anos estavam abaixo do percentil 50 para crianças saudáveis. A presença de sinais e sintomas de doença pulmonar aos três anos estava apenas fracamente associada ao crescimento e a categorias nutricionais nessa mesma idade. No entanto, os pacientes com menor crescimento e índices nutricionais aos três anos tinham função pulmonar inferior aos seis anos e esse foi evidente para P/I e A/I, em que as diferenças na porcentagem prevista de valores de função pulmonar atingiram 15 e 12 pontos para a mais baixa e mais alta categorias de P/I e A/I, respectivamente.
Peterson et al.14 prospectivamente examinaram dados de 319 crianças de seis a oito anos. A mudança total de peso (kg / mês) entre primeiras e últimas visitas da criança foi examinada como um marcador de crescimento cumulativo durante dois anos. Na análise de regressão de medidas repetidas, os valores de VEF1 não variaram significativamente pela mudança de altura em pacientes com mudança de alto ou baixo peso. Nem a altura, nem a mudança na altura da primeira observação foram significativamente associadas com valores de VEF1, mas as crianças que tiveram um ganho de peso constante mostraram tendência a experimentar um maior aumento da VEF1 do que aquelas que perderam peso.
Assael et al.15 seguiram 163 pacientes, a fim de avaliar a relação entre o crescimento linear e gravidade da doença pulmonar. Esse estudo mostrou uma perda progressiva da VEF1 em pacientes com doença pulmonar leve e grave. Em pacientes com doença grave, o início do crescimento pré‐púbere e o pico ocorreram mais tarde e a uma velocidade ligeiramente menor em comparação com pacientes com doença leve. O pico puberal de pacientes graves ocorreu cerca de oito meses mais tarde do que em pacientes leves. Sua velocidade de pico foi inferior em 1,3 centímetro/ano (vs. doença leve) e 2 centímetros/ano (comparada com indivíduos saudáveis). Quando a idade, altura e velocidade de crescimento em todas as quatro metas de crescimento foram consideradas como um todo, as diferenças entre os pacientes com doença leve e grave foram altamente significativas.
Yen et al.,16 em um estudo observacional prospectivo, avaliaram 3.142 pacientes com FC e descobriram que VEF1% foi menor no grupo de pacientes com %P/I<10% aos quatro anos do que todos os outros grupos de %P/I, nunca alcançou um VEF1>80% ao longo do período de estudo. Esse estudo também descobriu que os pacientes no maiores percentis de peso e altura aos quatro anos tiveram menos exacerbações pulmonares, passaram menos dias no hospital e tiveram melhor sobrevida aos 18 anos.
Woestenenk et al.17 estudaram 156 crianças em um período médio de 7,4 anos e não encontraram correlação entre VEF1% e peso ou altura, em uma primeira análise transversal. No entanto, independentemente da sua categoria inicial de P/I ou P/A, as crianças que tiveram aumento de peso apresentaram pequenos declínios no VEF1% do previsto de seis a 10 anos. O declínio do VEF1% desacelerou de 1,8% e 1,9%, respectivamente, para cada unidade de aumento na P/I e P/A. O estudo não encontrou qualquer associação entre VEF1 e A/I ou AI/AA.
Sanders et al.18 estudaram prospectivamente 6.805 pacientes no Cystic Fibrosis Foundation Patient Registry. As crianças com FC nascidas entre 1994 e 2005, seguidas de ≤2 anos até sete anos, foram avaliadas de acordo com mudanças nos percentis PC anualizados entre 0 e dois anos e os percentis de IMC entre dois e seis anos. Os resultados de um modelo de regressão linear multivariado mostraram que indivíduos com PC‐IMC sempre>percentil 50 apresentavam o maior VEF1% previsto aos seis a sete anos, significativamente maior do que os indivíduos cujos PC‐IMC era estável ou diminuiu>0 pontos percentuais.
DiscussãoEstudos direcionados para o manejo dos pacientes com FC têm contribuído para o avanço no atendimento multidisciplinar, com vistas ao melhor tratamento dessa doença. Apesar dessas melhorias, a insuficiência pulmonar permanece como a principal causa de morte na FC e há fortes associações entre o crescimento e índices nutricionais e função pulmonar.19 No entanto, a maioria dos estudos que relata essas evidências são de corte transversal, com poucas evidências para apoiar a associação entre alterações longitudinais no crescimento e nutrição com alterações na função pulmonar durante a vida.
O estudo feito por Assael et al.15 foi o primeiro a demonstrar uma clara associação entre eventos de crescimento inicial e função pulmonar em pacientes com FC. Esse estudo mostrou uma perda progressiva da VEF1 em pacientes com doença pulmonar leve e grave. Em pacientes com doença pulmonar grave, o início e o pico pré‐púberes ocorreram mais tarde e a uma velocidade ligeiramente menor em comparação com pacientes com doença leve. O pico puberal de pacientes graves ocorreu cerca de oito meses mais tarde do que em pacientes com doença leve. Sua velocidade de pico foi menor em 1,3 centímetro/ano (vs. doença leve) e 2 centímetros/ano (comparada com indivíduos saudáveis). Quando a idade, altura e velocidade de crescimento em todas as quatro metas de crescimento foram consideradas como um todo, as diferenças entre os pacientes com doença leve e grave foram altamente significativas. A principal conclusão desse estudo é que a baixa velocidade de crescimento é uma manifestação precoce da gravidade da doença pulmonar na FC.
Dois estudos14,17 não encontraram associações entre mudanças na altura e mudanças no VEF1, mas foram feitos em períodos mais curtos e, diferentemente dos dois estudos mencionados anteriormente,15,16 sua análise incluiu crianças entre dois a 10 anos. Assim, a influência de picos de crescimento não pôde ser avaliada. No entanto, ambos os estudos apontam para uma associação entre ganho de peso e melhor função pulmonar.
Três estudos12,13,18 relataram que a melhoria no crescimento e nos parâmetros clínicos nos primeiros anos de vida estava associada a uma melhor função pulmonar dois a cinco anos mais tarde. Assim, sugere‐se que o crescimento e o estado nutricional na infância podem ser fortes preditores da gravidade da doença pulmonar mais tarde na vida, o que é reforçado por Yen et al.,16 que mostraram que os pacientes nos percentis mais altos de peso e altura aos quatro anos tinham menos exacerbações pulmonares, passavam menos dias no hospital e tinham melhor sobrevida aos 18 anos.
Anormalidades na função pulmonar, incluindo diminuição do pico de fluxo e menor capacidade vital, capacidade vital forçada e fluxo expiratório forçado ocorrem em outras condições em que as crianças estão desnutridas e têm crescimento atrasado.20–23 Intervenções nutricionais com pacientes desnutridos mostraram que o aumento da ingestão calórica resultou em aumento das velocidades de ganho de peso e altura e melhorias em outras medidas de estado nutricional, além de redução nos casos de infecções pulmonares ou declínio mais lento da função pulmonar.10,24–26 Crescimento e nutrição são duas características intimamente ligadas; por isso, uma vez que uma melhor nutrição está associada a uma melhor função pulmonar em FC, a manutenção de um estado nutricional saudável é importante, não só para a nutrição e o crescimento, mas para a função pulmonar também.27 Stephenson et al.28 estudaram uma coorte de 909 pacientes com FC. Os indivíduos nas categorias sobrepeso e obesidade eram mais velhos e apresentavam melhor função pulmonar. Dentro do grupo de baixo peso, um aumento de 10% no IMC resultou em um aumento relativo de 4% no VEF1 e os indivíduos com IMC na faixa adequada tiveram um aumento relativo de 5% no VEF1. Esse foi o primeiro estudo que caracterizou mudanças no estado nutricional ao longo do tempo e quantificou a relação entre nutrição e função pulmonar em todo o espectro de categorias de IMC. Em relação à melhoria do crescimento, as evidências de ensaios clínicos também são limitadas. Terapia com hormônio de crescimento não parece melhorar a função pulmonar em um nível significativo.29
Em resumo, os estudos destacam a importância de melhorar o crescimento e o estado nutricional de pacientes com FC por meio de uma intervenção nutricional agressiva e também pelo tratamento da doença pulmonar, mesmo em pacientes com doença pulmonar leve. No entanto, a relação entre o crescimento longitudinal e a função pulmonar em FC ainda não está clara e deve ser mais amplamente investigada. Os ensaios clínicos controlados com terapias direcionadas para doenças pulmonares são necessários para investigar sua relação com o crescimento e o estado nutricional. Considerando a deterioração que os pulmões sofrem com o tempo na FC, estudos de longo prazo são importantes para melhor caracterizar esses parâmetros e identificar quais fatores estão mais profundamente envolvidos nesse processo e, assim, intervenções adequadas podem ser implantadas. Medidas mais sensíveis para a função pulmonar, tais como o índice de clearance pulmonar, também são necessárias para melhor diagnosticar a gravidade da doença pulmonar, principalmente na faixa etária pré‐escolar.30 Há cerca de 2.000 mutações do gene CFTR, distribuídas em seis classes. Assim, os estudos de associação entre as variáveis devem ser estruturados para as classes de mutação que configuram a doença mais grave (classes I, II e III). A maior facilidade de acesso aos medicamentos, atendimento em centros de referência, triagem neonatal, tratamento precoce de infecção por Pseudomonas aeruginosa, ingestão precoce de enzimas pancreáticas, o conhecimento de polimorfismos e seguimento diferenciado na adolescência e na idade adulta (principalmente para o sexo feminino) são fatores que podem equilibrar e modificar a história natural da FC no que diz respeito ao declínio da função pulmonar, crescimento e à nutrição desses pacientes.
FinanciamentoO estudo não recebeu financiamento.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Agradecemos a www.laboratoriomultiusuario.com.br e ao Laboratório de Fisiologia Pulmonar (Lafip) do Centro de Investigação em Pediatria (Ciped) pela ajuda na busca.
