



El síndrome de déficit del transportador de glucosa cerebral (GLUT1DS) puede presentar fenotipos variados, incluyendo epilepsia, déficit intelectual y trastorno del movimiento. La mayoría presenta hipoglucorraquia y/o defectos en el gen SLC2A1, aunque existen pacientes sin hipoglucorraquia y otros con genética de SLC2A1-negativa, o con defectos en otros genes y fenotipo compatible.
ObjetivosDescribir las características clínicas, bioquímicas y genéticas y realizar un análisis univariante de un grupo de pacientes con fenotipo clínico y bioquímico de GLUT1DS, con o sin genética SLC2A1-positiva.
Material y métodosSe incluyeron 13 pacientes con criterios clínico-bioquímicos de GLUT1DS. Se realizó secuenciación de SLC2A1 y MLPA. En los casos negativos se realizó exoma clínico.
ResultadosSeis presentaron fenotipo clásico, 2 discinesia paroxística, 2 trastornos del movimiento complejo, 2 ausencias precoces y otro presentó epilepsia con ausencias infantiles refractaria a farmacoterapia. Seis fueron SLC2A1-positivos. Y en 5 de los SLC2A1-negativos se identificó otro defecto genético. No hubo diferencias significativas entre los dos grupos en edad de inicio, presentación clínica, microcefalia, discapacidad intelectual ni respuesta a dieta cetogénica. De forma no significativa, los pacientes SCL2A1-positivos presentaron más cambios clínicos en relación con la ingesta (66,7% vs. 28,6%) y mayor persistencia de síntomas motores (66% vs. 28,6%). De forma significativa, presentaron menor glucorraquia (34,5mg/dl vs. 46mg/dl, p=0,04) e índice glucorraquia/glucemia más bajo (0,4 vs. 0,48, p=0,05) que los SLC2A1-negativos.
ConclusionesGLUT1DS puede ser causado por defectos genéticos en otros genes diferentes de SLC2A1 en pacientes con fenotipo compatible, hipoglucorraquia y buena respuesta a dieta cetogénica.
Glucose transporter type 1 (GLUT1) deficiency syndrome may present a range of phenotypes, including epilepsy, intellectual disability, and movement disorders. The majority of patients present low CSF glucose levels and/or defects in the SLC2A1 gene; however, some patients do not present low CSF glucose or SLC2A1 mutations, and may have other mutations in other genes with compatible phenotypes.
AimsWe describe the clinical, biochemical, and genetic characteristics of the disease and perform a univariate analysis of a group of patients with clinical and biochemical phenotype of GLUT1 deficiency syndrome, with or without SLC2A1 mutations.
Material and methodsThe study included 13 patients meeting clinical and biochemical criteria for GLUT1 deficiency syndrome. SLC2A1 sequencing and multiplex ligation-dependent probe amplification were performed; exome sequencing was performed for patients with negative results.
ResultsSix patients presented the classic phenotype; 2 paroxysmal dyskinesia, 2 complex movement disorders, 2 early-onset absence seizures, and one presented drug-resistant childhood absence epilepsy. Six patients were positive for SLC2A1 mutations; in the other 5, another genetic defect was identified. No significant differences were observed between the 2 groups for age of onset, clinical presentation, microcephaly, intellectual disability, or response to ketogenic diet. Patients with SLC2A1 mutations presented more clinical changes in relation to diet (66.7% vs. 28.6% in the SLC2A1-negative group) and greater persistence of motor symptoms (66% vs. 28.6%); these differences were not statistically significant. Significant differences were observed for CSF glucose level (34.5 vs. 46mg/dL, P=.04) and CSF/serum glucose ratio (0.4 vs. 0.48, P<.05).
ConclusionsGLUT1 deficiency syndrome may be caused by mutations to genes other than SLC2A1 in patients with compatible phenotype, low CSF glucose level, and good response to the ketogenic diet.
El síndrome de déficit del transportador de glucosa de tipo 1 (GLUT1DS) es una enfermedad neurometabólica tratable, causada por un defecto en el transporte de dicho sustrato a través de la barrera hematoencefálica1,2. Se evidencia en un descenso en la concentración de glucosa en el líquido cefalorraquídeo (LCR) no atribuible a otras causas y/o en un descenso en la cantidad o actividad de la proteína (GLUT1). Con frecuencia, esta situación conduce a manifestaciones clínicas por disregulación o aumento de la excitabilidad neuronal2 que se traduce en epilepsia típicamente precoz y refractaria a fármacos. El fenotipo clásico asocia además retraso del desarrollo psicomotor o discapacidad intelectual y/o microcefalia o trastorno del movimiento. La dieta cetogénica (DC) es la terapia de elección en estos pacientes, que pueden mostrar respuestas claramente favorables a la misma, especialmente en lo que se refiere a crisis epilépticas y síntomas motores. Si bien, existe variabilidad en la respuesta entre diferentes pacientes y esta suele ser menor en general para los síntomas cognitivos2. La mayoría de los casos se producen por la existencia de una mutación en el gen SLC2A1 (1p34.2), que codifica para el GLUT1 y conduce a una pérdida de función parcial del mismo (haploinsuficiencia)2. Este es el único defecto genético asociado a la enfermedad hasta el momento actual2,3. Existe un grupo de pacientes en los que el análisis genético molecular de SLC2A1 no detecta alteraciones, siendo difíciles de distinguir en el resto de las características clínico-bioquímicas de los positivos2.
En los últimos años se ha producido una expansión de fenotipos clínicos atípicos3,4 en los que debería sospecharse GLUT1DS, tales como epilepsia generalizada (sobre todo ausencias precoces, epilepsia mioclónico-atónica o epilepsia generalizada familiar), retraso del desarrollo psicomotor (RPM), trastorno del movimiento complejo (TMC), episodios paroxísticos desencadenados por ejercicio o ayuno, o una combinación de estos síntomas5. La respuesta inmediata a la DC podría apoyar el diagnóstico, así como el hallazgo de mejoría en registros electroencefalográficos (EEG) posprandiales respecto a los realizados en situación preprandial. La hipoglucorraquia llamativa que acompaña a las formas clásicas no siempre está presente en estas formas atípicas, en las que el estudio genético positivo para SLC2A1 permite el diagnóstico definitivo4-6.
En cuanto a los pacientes con análisis genético-molecular negativo para SLC2A1 y características clínicas y bioquímicas compatibles, la captación de glucosa en los eritrocitos sería otro parámetro confiable para el diagnóstico1,7, si bien su accesibilidad y empleo en la práctica clínica habitual es excepcional3. Otra prueba complementaria útil es la tomografía por emisión de positrones con fluorodesoxiglucosa (PET-FDG), que revela típicamente hipometabolismo en la corteza cerebral, sobre todo en región temporal medial y tálamo, con relativa preservación de los ganglios basales. Este patrón es constante, sin importar edad, historia de crisis, gravedad de la enfermedad o tratamiento8. Así mismo, los estudios EEG con registro preprandial y posprandial pueden ayudar en el proceso diagnóstico3,9. Si el registro en ayunas presenta anomalías, el registro posprandial podría (no siempre) mostrar mejoría de estas, indicando un fallo energético cerebral reversible causado por el defecto en GLUT13.
Por otro lado, en los últimos años se ha expandido rápidamente el espectro de mutaciones conocidas en SLC2A14,10 y además se hipotetiza que algunos de los casos con características clínicas y bioquímicas compatibles y estudio genético negativo podrían deberse a fallos en el ensamblaje, plegamiento, transporte a la membrana celular o activación del GLUT111,12. Así mismo, hasta el momento no se ha encontrado una clara relación genotipo-fenotipo que explique la variabilidad clínica que caracteriza a esta enfermedad (incluso entre familiares afectados), y tampoco se encuentra explicación para la variabilidad en la respuesta a la DC12,13. Se piensa que las diferencias podrían deberse a mecanismos adicionales como genes modificadores y proteínas que contribuyan a la fisiopatología de esta entidad compleja3,13.
En el contexto referido, reportamos una serie de pacientes con características clínicas y analíticas compatibles con GLUT1DS, con variabilidad en cuanto al resultado en el análisis genético molecular de SLC2A1. Realizamos un estudio comparativo entre pacientes con estudio genético positivo y negativo para este gen, con el objetivo de establecer posibles diferencias en cuanto a características clínicas, valores analíticos y/o respuesta a terapias, con especial atención a la DC. Igualmente se describen otras causas genéticas que podrían ocasionar, dentro de su espectro de posibles manifestaciones, un fenotipo clínico y bioquímico compatible con GLUT1DS, ya sea típico o atípico.
Material y métodosSe realizó un estudio retrospectivo de una muestra de pacientes con características clínicas y bioquímicas compatibles con GLUT1DS, estudiados en la unidad de enfermedades metabólicas y neurodegenerativas de un hospital de tercer nivel, entre los años 2002 y 2018.
Recogida de datos genotípicosSe realizó, en un laboratorio de referencia, un análisis molecular genético en el DNA obtenido mediante extracción automática de una muestra de sangre periférica. Se procedió a amplificar por PCR y secuenciar los exones que componen el gen SLC2A1 que codifica para el GLUT1 y las zonas intrónicas adyacentes al inicio y al final de cada uno de los exones. La secuenciación se realizó con un secuenciador automático (ABI 3130) y los electroferogramas se compararon con la secuencia de referencia (ENST00000426263-9). En los casos con resultado negativo se amplió el estudio mediante secuenciación masiva del exoma clínico. Las variantes patogénicas identificadas se confirmaron por secuenciación Sanger en el paciente y sus padres.
Recogida de datos fenotípicosSe recogieron las características epidemiológicas (sexo, edad) y los antecedentes familiares de primer y segundo grado de clínica compatible con GLUT1DS. Se recogió información detallada acerca del inicio y evolución de la enfermedad, lo que permitió identificar el fenotipo de presentación clínica en cada paciente. Se distinguieron un fenotipo clásico (FC) y un fenotipo atípico (FA), estableciéndose diferentes subclasificaciones dentro de ambos. El FC fue definido por presencia de la combinación de epilepsia refractaria y retraso psicomotor. Se distinguieron 2 subgrupos según el inicio de las crisis: considerándose precoz el anterior a los 2 años (FC1) y tardío el posterior (FC2); y se especificó la asociación o no con TMC crónico, con o sin trastorno del movimiento paroxístico y con o sin microcefalia adquirida. En la categoría de FA se distinguieron 4 subgrupos (enumerados del 1 al 4) según el predominio clínico de TMC (FA1) especificándose con o sin retraso mental; discinesia paroxística inducida por ejercicio (DPIE) (FA2), especificándose con o sin epilepsia; epilepsia-ausencia precoz (FA3); o bien ninguno de los anteriores (FA4). En el último caso, si se trataba de otro fenotipo concreto potencialmente relacionado (epilepsia mioclónico-astática, temblor distónico, hemiplejía alternante u otro tipo de EPNE o epilepsia) se especificó.
En cuanto al fenotipo bioquímico, se recogieron variables de interés: glucemia, glucorraquia, índice glucorraquia/glucemia y concentración de lactato en LCR. Se realizó un análisis citológico y bioquímico básico del LCR. Las determinaciones se realizaron estando el paciente en ayunas de al menos 4-6 horas, siendo este el tiempo necesario para lograr un nivel de glucosa estable en el compartimento de LCR3. La glucemia se determinó inmediatamente antes de la realización de la punción lumbar, ya que una posible hiperglucemia de estrés en dicho contexto invalidaría la determinación si esta se realizara después del mismo3. Como puntos de corte para considerar estos valores sugestivos o compatibles con déficit de glucosa cerebral se emplearon los establecidos previamente para esta enfermedad6, que en pacientes con clínica compatible corresponden a un valor para la edad igual o inferior al percentil 10 para la glucorraquia e igual o inferior al percentil 25 para el índice glucorraquia/glucemia, junto con una concentración de lactato igual o inferior al percentil 90. La concentración normal de lactato es condición obligatoria en el GLUT1DS, ya que actúa como discriminante de otras causas de hipoglucorraquia como la meningitis bacteriana o tuberculosa y ciertos errores congénitos del metabolismo6.
Recogida de resultados de otras pruebas complementarias potencialmente útilesEn aquellos pacientes en quienes se realizó, se recogió el resultado de la PET-FDG, considerándose compatible con GLUT1DS en caso de ajustarse a descripciones previamente establecidas para esta patología8.
Así mismo, se recogieron los estudios neurofisiológicos comparativos realizados en ayunas y después de ingerir alimentos ricos en glúcidos, considerándose positivos aquellos en los que se objetivó clara mejoría en el periodo posprandial respecto al preprandial, ya fuera en la calidad de la actividad de fondo como en la presencia o intensidad de anomalías intercríticas o de crisis epilépticas. El estudio se realizó en dichos casos mediante monitorización video-electroencefalográfica (VEEG).
Evolución en el tiempo y respuesta a la dieta cetogénica (DC)Se recogió, en caso oportuno, el número de fármacos antiepilépticos (FAE) recibidos y la eficacia en cuanto a control de las crisis epilépticas. Se especificó la administración o no de DC, tipo y tiempo durante el cual se realizó. Se excluyeron los casos en que el cumplimiento no fue adecuado o que por diferentes razones no alcanzaron niveles de cetonemia suficientes. En los casos con cumplimiento y cetonemias evolutivas adecuadas se especificó la eficacia o no de la DC de forma global y en concreto en 3 aspectos: control de crisis, control de anomalías motoras, y mejoría en rendimiento escolar u otros aspectos cognitivos. Se consideró eficacia de la DC (o mejoría significativa) tanto el control completo de las crisis (desaparición) como el control parcial de las mismas, entendido como la reducción de su frecuencia en más del 50% respecto a la inicial. Así mismo, se tuvo en cuenta la consiguiente retirada de FAE permitida por el control mantenido de las crisis epilépticas. En cuanto al control de síntomas motores, en aquellos de curso episódico (DPIE o TMC con síntomas paroxísticos) se consideró mejoría significativa la desaparición (control completo) o bien la reducción de la frecuencia de episodios en más del 50% (control parcial). En aquellos con clínica motora permanente (consistente principalmente en dificultades en la coordinación, equilibrio y/o ataxia…) la DC se consideró eficaz en los casos en que la mejoría tras su inicio permitió la consecución de avances motores objetivados en controles clínicos (inicio o recuperación de la marcha o mejoría del patrón de esta, mejor ejecución de tareas de la vida diaria que requieren coordinación). En cuanto al aspecto cognitivo, en niños con discapacidad intelectual moderada-grave se valoró la aparición de avances en la adquisición de aprendizajes (con mayor velocidad o complejidad que previamente…) tras la instauración de la DC. Del mismo modo, se valoró positivamente la mejoría en atención, comunicación-interacción o lenguaje. Para ello se recogió la información referida en reportes psicopedagógicos y/o escolares, y lo referido por la familia y objetivado por observación clínica en sucesivos controles médicos. En niños cognitivamente normales o con discapacidad intelectual leve se consideró respuesta a la DC la mejoría en aspectos relacionados con la atención, razonamiento y rendimiento académico en relación con el inicio de esta. La obtención de estos datos se llevó a cabo generalmente mediante revisión de informes psicopedagógicos, escolares y a veces determinadas pruebas neuropsicológicas, junto con la entrevista a la familia y la observación y exploración del paciente en los controles médicos.
En cuanto a la evolución clínica, en todos los casos se recogió la aparición o no, durante el seguimiento, de afectación cognitiva y su gravedad, microcefalia, trastorno de movimiento, y/o alteración específica en las funciones ejecutivas. Las capacidades cognitivas y las alteraciones en funciones específicas se determinaron mediante la realización de baterías de escalas y test neuropsicológicos incluyendo la determinación del Coeficiente Intelectual (CI) mediante la escala de inteligencia de Wechsler para niños-IV (WISC-IV) o para preescolar y primaria (WPPSI-IV) según la edad. En aquellos cuya afectación impedía (generalmente por su gravedad o nula colaboración) su adecuada realización, el grado de disfunción se estimó mediante valoración clínica en una consulta especializada (neuropediatra y/o neuropsicólogo experto).
Análisis estadísticoLa homogeneidad de las variables demográficas, antecedentes médicos y otros parámetros clínicos fueron analizados. La descripción se realizó mediante la media, mediana, desviación típica de las variables cuantitativas, así como con la frecuencia absoluta y frecuencia relativa de las variables cualitativas. Para las variables cuantitativas se desarrollaron pruebas de t-Student en caso de cumplirse la asunción de normalidad y pruebas no paramétricas de U de Mann-Whitney en caso contrario. Las variables cualitativas fueron analizadas mediante test de homogeneidad basadas en la distribución χ2 cuando los valores esperados lo hicieron posible y mediante test exacto de Fisher en caso contrario. Se consideró significación estadística valores de p<0,05. Se analizaron mediante el paquete estadístico SPSS 22.0 IBM Inc.
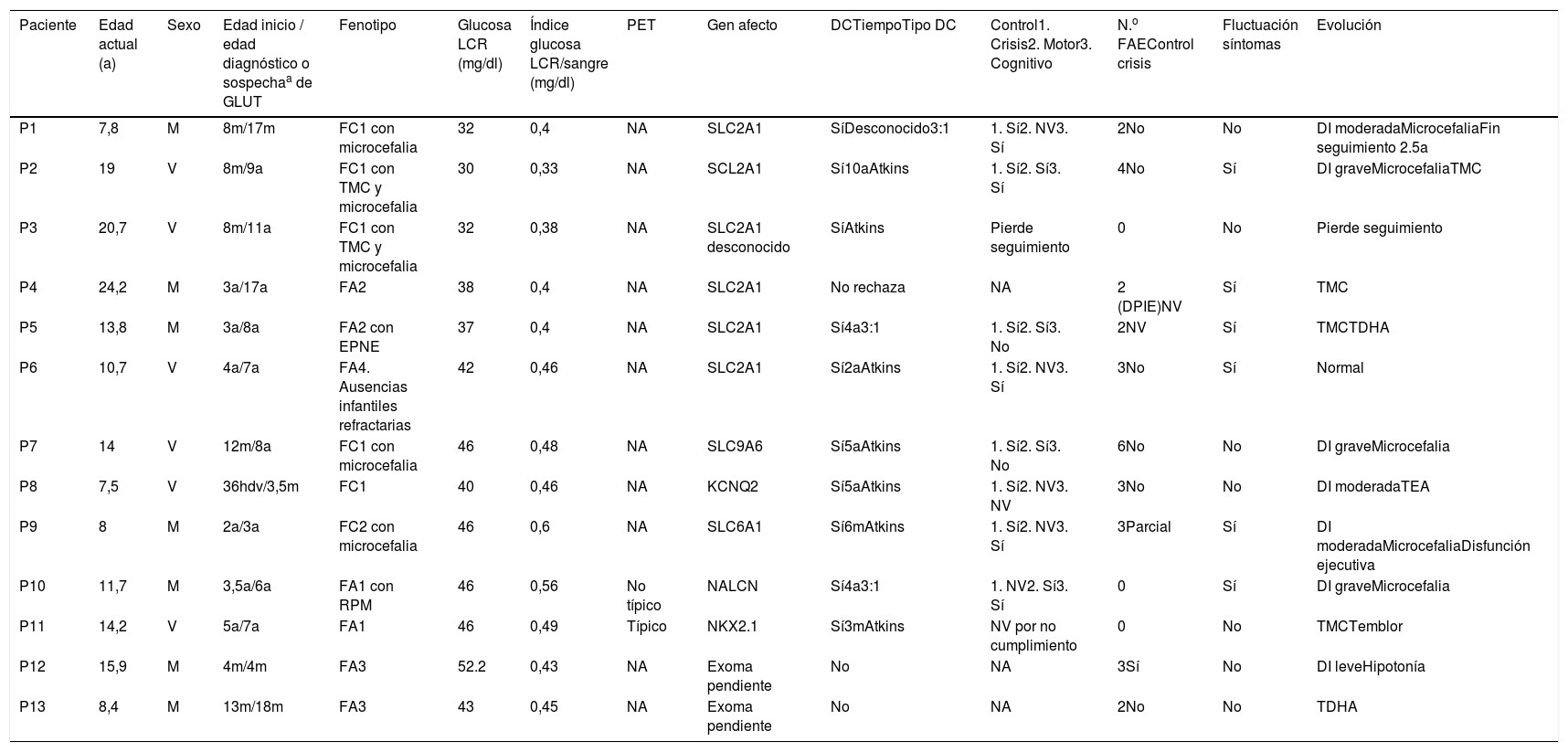
Resultados (tabla 1)Se incluyeron 13 pacientes (6 varones, 7 mujeres), seguidos durante un periodo de entre 22 meses y 14 años. Solo 2 pacientes presentaban antecedentes familiares atribuibles a GLUT1DS, ambos con genética positiva.
Tabla de pacientes
| Paciente | Edad actual (a) | Sexo | Edad inicio / edad diagnóstico o sospechaa de GLUT | Fenotipo | Glucosa LCR (mg/dl) | Índice glucosa LCR/sangre (mg/dl) | PET | Gen afecto | DCTiempoTipo DC | Control1. Crisis2. Motor3. Cognitivo | N.o FAEControl crisis | Fluctuación síntomas | Evolución |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | 7,8 | M | 8m/17m | FC1 con microcefalia | 32 | 0,4 | NA | SLC2A1 | SíDesconocido3:1 | 1. Sí2. NV3. Sí | 2No | No | DI moderadaMicrocefaliaFin seguimiento 2.5a |
| P2 | 19 | V | 8m/9a | FC1 con TMC y microcefalia | 30 | 0,33 | NA | SCL2A1 | Sí10aAtkins | 1. Sí2. Sí3. Sí | 4No | Sí | DI graveMicrocefaliaTMC |
| P3 | 20,7 | V | 8m/11a | FC1 con TMC y microcefalia | 32 | 0,38 | NA | SLC2A1 desconocido | SíAtkins | Pierde seguimiento | 0 | No | Pierde seguimiento |
| P4 | 24,2 | M | 3a/17a | FA2 | 38 | 0,4 | NA | SLC2A1 | No rechaza | NA | 2 (DPIE)NV | Sí | TMC |
| P5 | 13,8 | M | 3a/8a | FA2 con EPNE | 37 | 0,4 | NA | SLC2A1 | Sí4a3:1 | 1. Sí2. Sí3. No | 2NV | Sí | TMCTDHA |
| P6 | 10,7 | V | 4a/7a | FA4. Ausencias infantiles refractarias | 42 | 0,46 | NA | SLC2A1 | Sí2aAtkins | 1. Sí2. NV3. Sí | 3No | Sí | Normal |
| P7 | 14 | V | 12m/8a | FC1 con microcefalia | 46 | 0,48 | NA | SLC9A6 | Sí5aAtkins | 1. Sí2. Sí3. No | 6No | No | DI graveMicrocefalia |
| P8 | 7,5 | V | 36hdv/3,5m | FC1 | 40 | 0,46 | NA | KCNQ2 | Sí5aAtkins | 1. Sí2. NV3. NV | 3No | No | DI moderadaTEA |
| P9 | 8 | M | 2a/3a | FC2 con microcefalia | 46 | 0,6 | NA | SLC6A1 | Sí6mAtkins | 1. Sí2. NV3. Sí | 3Parcial | Sí | DI moderadaMicrocefaliaDisfunción ejecutiva |
| P10 | 11,7 | M | 3,5a/6a | FA1 con RPM | 46 | 0,56 | No típico | NALCN | Sí4a3:1 | 1. NV2. Sí3. Sí | 0 | Sí | DI graveMicrocefalia |
| P11 | 14,2 | V | 5a/7a | FA1 | 46 | 0,49 | Típico | NKX2.1 | Sí3mAtkins | NV por no cumplimiento | 0 | No | TMCTemblor |
| P12 | 15,9 | M | 4m/4m | FA3 | 52.2 | 0,43 | NA | Exoma pendiente | No | NA | 3Sí | No | DI leveHipotonía |
| P13 | 8,4 | M | 13m/18m | FA3 | 43 | 0,45 | NA | Exoma pendiente | No | NA | 2No | No | TDHA |
a: años; D: día; DC: dieta cetogénica; DI: discapacidad intelectual; DPIE: discinesia paroxística inducida por ejercicio; EPNE: episodios paroxísticos no epilépticos; FAE: fármacos antiepilépticos; hdv: horas de vida; LCR: líquido cefalorraquídeo; m: meses; M: mujer; NA: no aplicable; NV: no valorable; PET: tomografía por emisión de positrones; RPM: retraso del desarrollo psicomotor; TDAH: trastorno por déficit de atención e hiperactividad; TEA: trastorno del espectro autista; TMC: trastorno del movimiento complejo V: varón.
El análisis genético molecular de SLC2A1 detectó cambios patológicos en 5 pacientes. A efectos de análisis, se incluye en el mismo grupo un sexto paciente con resultado desconocido, diagnosticado con base en valores de glucorraquia menores de 33mg/dl en 2 determinaciones distintas junto con fenotipo clínico típico. De los 7 restantes, el estudio genético ampliado mostró algún cambio patogénico en 5 pacientes, afectando en 4 de ellos a genes codificantes de otros canales o transportadores diferentes de GLUT1.
Datos fenotípicos (clínicos y bioquímicos): análisis comparativoLa edad media de inicio clínico fue de un año (rango 0,01-5), sin diferencias entre pacientes con estudio genético positivo y negativo para SLC2A1. Tampoco se objetivaron diferencias en cuanto a predominio de sexo. El fenotipo clásico se presentó en 6 casos, 5 con epilepsia de inicio precoz (FC1) y el restante a los 2 años y 7 meses (FC2) en forma de ausencias precoces con retraso psicomotor. Los casos con fenotipo atípico se distribuyeron del siguiente modo: 2 presentaban TMC (FA1) —uno de ellos con afectación cognitiva—, 2 DPIE (FA2), 2 epilepsia-ausencia de inicio precoz (FA3), y otro presentó epilepsia tipo ausencias infantiles de difícil control, no englobable en el resto de categorías definidas (FA4).
No se encontraron diferencias significativas entre los 2 grupos respecto a la edad de inicio de la clínica (2,1 vs. 1,9, p=0,8), forma clínica de presentación (fenotipo clásico 50% vs. 42,8%) ni presencia de microcefalia (50% vs. 42,8%), ni en la evolución con discapacidad intelectual ni su gravedad. La discapacidad intelectual se dio de forma evolutiva en todos aquellos casos cuyo fenotipo inicial incluía retraso psicomotor (todos los pacientes con FC y un paciente con FA1), siendo la afectación moderada-grave. Los casos restantes, con desarrollo psicomotor inicial normal, no presentaron deterioro cognitivo durante el seguimiento. No se encontraron diferencias significativas en la evolución con disfunción ejecutiva (presente en 5 pacientes) que solo se contempló en 7 pacientes, siendo no valorable en el resto por discapacidad intelectual grave.
Aunque de forma no significativa, los pacientes con genética positiva para SCL2A1 presentaron fluctuación de los síntomas tras la ingesta de forma más frecuente (66,7% vs. 28,6%, p=0,28) y una mayor persistencia de síntomas motores durante el seguimiento (66% vs. 28,6%, p=0,29).
En cuanto a las características bioquímicas, de forma estadísticamente significativa, los pacientes SCL2A1 positivos presentaban glucorraquia más baja (34,5mg/dl [30-42] vs. 46mg/dl [40-52], p=0,04) y un índice glucosa LCR/sangre más bajo (0,4 [0,3-0,46] vs. 0,48 [0,43-0,6], p=0,05). El estudio citológico y bioquímico no mostró otros hallazgos significativos.
Datos fenotípicos: análisis descriptivo por gruposDe los pacientes SLC2A1 positivos, 3 presentaban fenotipo clínico clásico (P1, P2 y P3), todos con epilepsia de inicio precoz y microcefalia, 2 de ellos asociaban además TMC. Estos 3 pacientes presentaron los valores de glucorraquia más bajos de la serie, presentando todos un valor igual o inferior a 32mg/dl. Los otros 3 pacientes SLC2A1 positivos (P4, P5 y P6) presentaban fenotipo atípico, 2 de ellos tipo DPIE, ambos sin epilepsia y sin afectación cognitiva (P4 y P5). El P6 presentó clínica de ausencias infantiles de difícil control, con antecedente de clínica similar en un tío materno; detectándose la misma mutación en su madre (que ha permanecido asintomática en todo momento). Los pacientes SLC2A1 con fenotipo atípico comenzaron con clínica de forma más tardía que los típicos y presentaron valores mayores de glucorraquia.
De los pacientes SLC2A1 negativos con otra alteración genética, 3 presentaron fenotipo clínico clásico (P7, P8 y P9). El P7 presentó epilepsia de inicio precoz (12 meses) junto con retraso psicomotor y microcefalia y cuya epilepsia mejoró significativamente tras inicio de DC (no así el aspecto cognitivo). El análisis genético mediante secuenciación masiva identificó en hemicigosis una mutación nueva presumiblemente severa en el gen SLC9A6 (c.803+1G>A), que codifica para el transportador de sodio/protones SLC9A6, cuya deficiencia causa el síndrome de Christianson14 (retraso mental grave ligado al cromosoma X (Xq26.3), ausencia de lenguaje, TEA, epilepsia, microcefalia, ataxia de comienzo tardío, debilidad y distonía). El estudio genético de la madre confirmó la misma mutación. El P8 presentó epilepsia de inicio neonatal refractaria a politerapia farmacológica y respuesta completa inmediata a DC (6 años libre de crisis actualmente). Desarrolló evolutivamente un trastorno del espectro autista. En este paciente se detectó en heterocigosis una variante alélica previamente descrita como patogénica (c.619C>T, p.Arg207Trp) en el gen KCNQ2 (20q13.33), descrito en la epilepsia neonatal benigna y más recientemente en un espectro de encefalopatías epilépticas de inicio precoz15. En el P9, la epilepsia comenzó a los 2 años y 7 meses con episodios de desviación de la mirada vertical sin aparente desconexión inicial. Evolucionó con ausencias que fueron detectadas en un registro VEEG en ayunas objetivándose su desaparición en el registro unos 30 minutos tras ingesta de azúcares junto con una disminución clara de las anomalías epileptiformes. Se identificó una variante patogénica de novo (c.T277delGC, p.Ala93Glyfs*113) en el gen SLC6A1 (3p25.3) no descrita en las bases de datos genómicas; que se predice con efecto patogénico por conllevar la síntesis de una proteína truncada. El gen SLC6A1 codifica para un transportador de ácido gamma-aminobutírico (GABA) localizado en la membrana plasmática, y se ha asociado con epilepsia mioclónica-astática o síndrome de Doose16.
Los otros 2 pacientes con estudio SLC2A1 negativo y otra mutación detectada en el exoma (P10 y P11) presentaron fenotipo atípico. El P10, con antecedente de retraso psicomotor desde las primeras fases, debutó con episodios distónicos de distribución cervical y miembros superiores, y evolucionó a TMC con ataxia, distonía y temblor. Presentaba además episodios de hipoactividad de difícil caracterización que mejoraban con la ingesta. Se inició DC (ratio inicial 3:1) con mejoría llamativa de los episodios distónicos y mejoría global del trastorno motor y el rendimiento cognitivo, situación que mantiene actualmente (5 años después) con dieta de bajo índice glucémico. En este paciente se detectó una mutación de novo (p.Ile322Thr) en el gen NALCN (13q32.3-q33.1) codificante de un canal iónico cuyo defecto se asocia a síndrome de CLIFHADD17,18(contractures of limbs and face, hypotonia, developmental delay). El P11 comenzó con torpeza y alteración del patrón de marcha más notoria desde los 3 años, con evolución a TMC con predominio de distonía y temblor, sin afectación cognitiva y tratado con levotiroxina desde siempre. Se realizó un panel de distonías con resultado negativo, y PET-FDG que mostró patrón compatible con GLUT1DS. El estudio mediante secuenciación masiva identificó en heterocigosis una variante alélica nueva (c.727delC, p.Arg243Alafs*4) en el gen NKX2-1 (14q33.3), no incluida en la base de datos profesional ni en las bases de datos poblacionales pero con predicciones bioinformáticas indicativas de patogenicidad. Este gen parece implicado en el desarrollo del prosencéfalo, glándula tiroides y pulmones durante el desarrollo embrionario y su alteración se asocia al síndrome de coreoatetosis e hipotiroidismo congénito con o sin disfunción respiratoria, de herencia autosómica dominante19.
Resultados de otras pruebas complementarias potencialmente útilesSolo en 2 pacientes se realizó como parte del estudio un PET-FDG (ambos con genética negativa para SCL2A1), siendo en uno de los casos compatible con GLUT1DS (P11) y en el otro no sugestivo (P10).
Respuesta a la dieta cetogénica (DC)Todos los casos con genética SLC2A1 positiva con crisis epilépticas mostraron refractariedad a FAE en politerapia, mientras que 3 de los 7 casos con genética SLC2A1 negativa mostraron respuesta evolutiva favorable con FAE aunque con difícil control inicial.
Diez de los 13 pacientes iniciaron DC, 8 de los cuales (4 de ellos SLC2A1 positivos) con buen cumplimiento y cetonemias adecuadas, durante un periodo de entre 6 meses y 8 años y 10 meses (mediana 4,5 años). Todos los pacientes SLC2A1 positivos mostraron mejoría significativa. Tres de ellos (P1, P2, P6) presentaban crisis epilépticas refractarias y frecuentes (entre diarias y mensuales) y el restante (P5) DPIE y EPNE de dudosa etiología (clínica poco compatible) pero con brotes de actividad paroxística generalizada en el EEG. Los pacientes P1 y P6 presentaron respuesta completa a la DC con desaparición precoz de las crisis tras su inicio. En el P2 la mejoría fue muy notable, presentando alguna crisis de forma muy ocasional. En los 3 pacientes, la espectacular mejoría permitió la retirada progresiva de FAE, pasando desde la politerapia (con combinaciones de 2 o 3 FAE) hasta prácticamente la suspensión completa. Si bien, en todos ellos las crisis reaparecieron tras la retirada del último FAE, por lo que finalmente se mantuvo, volviendo a la situación de control previa con DC y un solo FAE. Todos ellos presentaron mejoría en atención y/o capacidad de comunicación. En el caso del paciente P5 los episodios paroxísticos (tanto por su DPIE como los EPNE) desaparecieron tras el inicio de DC, así como la actividad paroxística en el EEG. A lo largo de la evolución ha presentado algún episodio de discinesia paroxística, muy aisladamente y en relación con ejercicio físico de muy elevada intensidad. En el grupo de pacientes SLC2A1 negativos, se objetivó mejoría en los 4 casos con mutaciones identificadas en genes codificantes de otros canales. El único paciente que no compartía esta característica abandonó la DC 3 meses tras su inicio por ineficacia, si bien el cumplimiento no fue óptimo de forma mantenida. De los que presentaron mejoría, 2 (P8 y P9) presentaban epilepsia refractaria con crisis pluridiarias que cedieron tras la instauración completa de la DC, asociada a FAE en ambos casos. El paciente restante (P10) presentaba episodios de distonía en ocasiones con rigidez de un hemicuerpo que disminuyeron en frecuencia hasta su desaparición. En los 2 últimos pacientes (P9 y P10) además se objetivó cierta mejoría en el rendimiento cognitivo y la atención.
La edad de inicio de la DC fue muy variable, entre los 4 meses de edad (P8) y 9,5 años (P2), no existiendo en nuestra muestra diferencias significativas atribuibles exclusivamente a la precocidad de su instauración.
DiscusiónLa complejidad y variabilidad creciente de fenotipos y genotipos asociados a GLUT1DS, así como la ausencia de una correlación clara genotipo-fenotipo, supone un reto en el diagnóstico y manejo terapéutico de los pacientes con criterios clínico-bioquímicos compatibles con esta entidad. Nuestro estudio refleja la realidad de esta complejidad, destacando la variabilidad fenotípica entre pacientes con estudio genético (SLC2A1) positivo, así como el solapamiento entre estos y los pacientes con estudio genético negativo que pueden ser indistinguibles y comportarse como verdaderas fenocopias2,9. Todo ello debe interpretarse teniendo en cuenta la limitación que supone un tamaño muestral relativamente bajo pero que por otro lado puede ser más representativo de aquello que podemos encontrar en la práctica clínica. De este modo, el solapamiento y la difícil distinción entre pacientes con genética positiva y negativa para SCL2A1 previamente descrita2,9 queda reflejada en los resultados obtenidos en el estudio comparativo de las características clínicas en nuestra muestra, que no mostraron diferencias significativas entre ambos grupos en cuanto a frecuencia de presentación con fenotipo clínico típico o atípico, microcefalia, o discapacidad intelectual (ni en su grado).
Se objetiva diferencia, aunque no significativa, en cuanto a la presencia de trastorno de movimiento (ya sea al inicio o durante la evolución) siendo su frecuencia superior en el grupo con genética positiva para SCL2A1 y destacando la DPIE, que solo se presentó en pacientes con estudio genético positivo para SLC2A1. Esta es una característica clínica bien documentada en individuos con GLUT1DS, y es una de las formas de presentación cuyo reconocimiento y detección ha aumentado en los últimos años, especialmente las formas sin epilepsia y con rendimiento cognitivo normal o levemente afectado2-4,19. El aumento de detección, así como de otros trastornos de movimiento no epilépticos (que a veces se consideran variantes menos graves de la enfermedad), despertó interés en la aplicación de la DC en este tipo de manifestaciones consideradas presumiblemente menos graves que la epilepsia pero que pueden llegar a ser incapacitantes2,4,20. En nuestro estudio se probó DC en todos los pacientes cuyo fenotipo clínico incluía TMC o DPIE, mejorando las anomalías motoras en todos los casos que demostraron adecuado cumplimiento (mediante controles de cetonemia) y por un tiempo suficiente. Llama la atención la respuesta objetivada en el caso SLC2A1 negativo con fenotipo de TMC con mutación de novo en el gen NALCN (causante del síndrome de Clifhadd). Así mismo, se objetivó diferencia, aunque no significativa, en la fluctuación de los síntomas o empeoramiento en relación con el ayuno, detectándose con más frecuencia en pacientes con genética clásica que en aquellos con una mutación alternativa. Aun así, 2 pacientes con genética SLC2A1 negativa mostraron esta fluctuación: el paciente con mutación en NACLN y el paciente con mutación en SLC6A1, que además mostró mejoría posprandial en VEEG.
En cuanto a la eficacia global de la DC (excluyendo los casos de mal cumplimiento o por tiempo insuficiente), todos los pacientes con genética positiva para SLC2A1 experimentaron mejoría clínica. Del grupo de pacientes con genética SLC2A1 negativa, todos los pacientes con diagnóstico alternativo de mutación en un gen codificante de un canal iónico mostraron mejoría global. En la práctica clínica actual no disponemos de parámetros predictivos de respuesta a DC2,12,13. Según estudios previos, el grado de respuesta a la misma es variable entre distintos individuos incluso compartiendo la misma mutación en SLC2A112,13. La mutación de SLC2A1 parece no ser el único factor y de hecho se postula en los casos negativos una posible influencia de factores postranscripcionales en la proteína codificada, así como la posible influencia de otros genes modificadores y proteínas que contribuyan a la fisiopatología de esta compleja entidad. La buena respuesta a DC y los cambios VEEG tras ingesta referidos en pacientes de nuestra muestra con estudio genético negativo para SLC2A1 y una mutación alternativa en algún gen codificante de otro canal podrían estar mediados por relaciones de este tipo, si bien no es posible afirmarlo ya que serían necesarios estudios bioquímicos exhaustivos.
Considerando lo anterior, el GLUT1DS debe tenerse en cuenta como posibilidad diagnóstica ante cualquiera de los fenotipos compatibles descritos. Esto permitiría un diagnóstico más precoz, así como la instauración de DC consiguiéndose en muchos casos una mejoría clínica notoria e incluso el control completo de alguno de los síntomas, siendo esto aplicable independientemente del resultado del estudio genético posterior (SLC2A1 positivo o negativo). En muchos casos facilitaría la suspensión o descenso del número de FAE necesarios para el control clínico e incluso podría evitar otros tratamientos más invasivos. Las características bioquímicas fueron las únicas que mostraron diferencias estadísticamente significativas, con valores de glucorraquia y de índice glucosa LCR/glucemia significativamente inferiores en los pacientes con estudio genético SLC2A1 positivo. Los casos de DPIE con cognitivo normal presentaron valores más altos, lo que coincide con otras descripciones previas4,6,10, aunque puede haber solapamiento frente a los pacientes con estudio SLC2A1 negativo.
ConclusionesLa complejidad y variabilidad creciente de fenotipos y genotipos asociados a déficit de GLUT1DS, así como la ausencia de una correlación clara genotipo-fenotipo, supone un reto en el diagnóstico y manejo terapéutico de pacientes con criterios clínico-bioquímicos compatibles con esta enfermedad; especialmente en casos en que no se detectan alteraciones en el gen SLC2A1.
FinanciaciónIdentificación y caracterización clínica y bioquímica de pacientes con síndrome GLUT1 (GLUT1DS): monitorización del tratamiento.” Proyectos de Investigación Traslacional 2017, CIBERER. OP: Dr. Luis González Gutiérrez-Solana (GCV6). Unidades participantes: U703 (Artuch); U746 (Pérez); GCV5 (Couce); GCV6 (Gutiérrez-Solana); GCV7 (López Laso); GCV8 (Del Toro). PdI: Medicina Metabólica Hereditaria.
Conflicto de interesesLos autores del trabajo expresan su conformidad con los contenidos del manuscrito y manifiestan la inexistencia de conflictos de interés.

