



Glucose transporter type 1 (GLUT1) deficiency syndrome may present a range of phenotypes, including epilepsy, intellectual disability, and movement disorders. The majority of patients present low CSF glucose levels and/or defects in the SLC2A1 gene; however, some patients do not present low CSF glucose or SLC2A1 mutations, and may have other mutations in other genes with compatible phenotypes.
AimsWe describe the clinical, biochemical, and genetic characteristics of the disease and perform a univariate analysis of a group of patients with clinical and biochemical phenotype of GLUT1 deficiency syndrome, with or without SLC2A1 mutations.
Material and methodsThe study included 13 patients meeting clinical and biochemical criteria for GLUT1 deficiency syndrome. SLC2A1 sequencing and multiplex ligation-dependent probe amplification were performed; exome sequencing was performed for patients with negative results.
ResultsSix patients presented the classic phenotype; 2 paroxysmal dyskinesia, 2 complex movement disorders, 2 early-onset absence seizures, and one presented drug-resistant childhood absence epilepsy. Six patients were positive for SLC2A1 mutations; in the other 5, another genetic defect was identified. No significant differences were observed between the 2 groups for age of onset, clinical presentation, microcephaly, intellectual disability, or response to ketogenic diet. Patients with SLC2A1 mutations presented more clinical changes in relation to diet (66.7%, vs 28.6% in the SLC2A1-negative group) and greater persistence of motor symptoms (66% vs 28.6%); these differences were not statistically significant. Significant differences were observed for CSF glucose level (34.5 vs 46mg/dL, P=.04) and CSF/serum glucose ratio (0.4 vs 0.48, P<.05).
ConclusionsGLUT1 deficiency syndrome may be caused by mutations to genes other than SLC2A1 in patients with compatible phenotype, low CSF glucose level, and good response to the ketogenic diet.
El síndrome de déficit del transportador de glucosa cerebral (GLUT1DS) puede presentar fenotipos variados, incluyendo epilepsia, déficit intelectual y trastorno del movimiento. La mayoría presentan hipoglucorraquia y/o defectos en el gen SLC2A1, aunque existen pacientes sin hipoglucorraquia y otros con genética de SLC2A1-negativa, o con defectos en otros genes y fenotipo compatible.
ObjetivosDescribir las características clínicas, bioquímicas y genéticas y realizar un análisis univariante de un grupo de pacientes con fenotipo clínico y bioquímico de GLUT1DS, con o sin genética SLC2A1-positiva.
Material y métodosSe incluyeron 13 pacientes con criterios clínico-bioquímicos de GLUT1DS. Se realizó secuenciación de SLC2A1 y MLPA. En los casos negativos se realizó exoma clínico.
ResultadosSeis presentaron fenotipo clásico, 2 discinesia paroxística, 2 trastornos del movimiento complejo, 2 ausencias precoces y otro presentó epilepsia con ausencias infantiles refractaria a farmacoterapia. Seis fueron SLC2A1-positivos. Y en 5 de los SLC2A1-negativos se identificó otro defecto genético. No hubo diferencias significativas entre los dos grupos en edad de inicio, presentación clínica, microcefalia, discapacidad intelectual ni respuesta a dieta cetogénica. De forma no significativa, los pacientes SCL2A1-positivos presentaron más cambios clínicos en relación con la ingesta (66,7% vs. 28,6%) y mayor persistencia de síntomas motores (66% vs. 28,6%). De forma significativa, presentaron menor glucorraquia (34,5mg/dl vs. 46mg/dl, p=0.04) e índice glucorraquia/glucemia más bajo (0,4 vs. 0,48, p=0,05) que los SLC2A1-negativos.
ConclusionesGLUT1DS puede ser causado por defectos genéticos en otros genes diferentes de SLC2A1 en pacientes con fenotipo compatible, hipoglucorraquia y buena repuesta a dieta cetogénica.
Glucose transporter type 1 (GLUT1) deficiency syndrome (GLUT1DS) is a treatable neurometabolic disorder caused by impaired glucose transport across the blood-brain barrier.1,2 These patients present low CSF glucose levels not attributable to other causes and/or a decrease in the concentration or activity of the GLUT1 protein. This frequently results in clinical manifestations of dysregulated or increased neuronal excitability,2 which typically causes early-onset refractory epilepsy. The classic phenotype also includes psychomotor developmental delay or intellectual disability, and/or microcephaly or movement disorders. Ketogenic dietary therapies (KDT) constitute the treatment of choice for these patients, achieving clear improvements, particularly with regard to seizures and motor symptoms. However, response varies from patient to patient and may be less marked in the case of cognitive symptoms.2 Most cases of GLUT1DS are caused by a mutation in SLC2A1 (located on chromosome 1p34.2), the gene that encodes GLUT1, leading to partial loss of protein function (GLUT1 haploinsufficiency).2 To date, this is the only genetic defect that has been associated with the disease.2,3 In some patients, genetic analysis of SLC2A1 does not detect alterations, despite the presence of similar clinical and biochemical characteristics to those observed in patients with positive results.2
In recent years, the clinical and phenotypic spectrum of GLUT1DS has expanded3,4 to include generalised epilepsy (especially early-onset absence epilepsy, myoclonic atonic epilepsy, and familial generalised epilepsy), psychomotor developmental delay, complex movement disorder (CMD), exercise- or fasting-induced paroxysmal dyskinesia, or a combination of these.5 Immediate response to KDT may support the diagnosis of GLUT1DS; improvements in EEG results after food intake, as compared to findings before eating, may also suggest GLUT1DS. Low CSF glucose levels constitute a typical finding in patients with classic forms of the disease, but are not always observed in atypical forms; in these cases, a definitive diagnosis is established if genetic studies detect a mutation in SLC2A1.4–6
In patients with negative molecular genetic study results for SLC2A1 mutations and compatible clinical and biochemical findings, erythrocyte glucose uptake represents a reliable diagnostic parameter,1,7 although erythrocyte glucose uptake assays are rarely available or used in clinical practice.3 Positron emission tomography with fluorodeoxyglucose (FDG-PET) is another useful complementary test; this imaging technique typically reveals hypometabolism in the cerebral cortex, particularly in the medial temporal region and thalamus, with the basal ganglia being relatively preserved. This pattern is consistently found in patients with GLUT1DS, regardless of age, history of seizures, disease severity, or treatment.8 EEG studies performed before and after food intake may also inform diagnosis.3,9 If EEG performed under fasting conditions detects abnormalities, a subsequent study performed after eating may reveal improvements, which would suggest reversible brain energy failure secondary to GLUT1 deficiency.3
The spectrum of SLC2A1 mutations causing GLUT1DS has expanded in recent years.4,10 It has also been hypothesised that some cases of compatible clinical and biochemical features with negative genetic results may be due to alterations in GLUT1 assembly, folding, membrane transport, or activation.11,12 To date, no clear genotype-phenotype relationship has been found to explain the clinical variability of the disease (even between affected individuals from the same family), and no explanation has been proposed for the variability in patient response to KDT.12,13 These differences are thought to be explained by other mechanisms, including modifier genes and proteins that may contribute to the pathophysiology of this complex entity.3,13
In the light of the above, we present a series of patients presenting clinical and laboratory findings compatible with GLUT1DS and variable results in molecular genetic studies of the SLC2A1 gene. We analysed clinical and laboratory parameters and treatment response, paying special attention to KDT, in order to compare patients with and without SLC2A1 mutations. We also describe other genetic factors potentially causing clinical and biochemical phenotypes compatible with GLUT1DS.
Material and methodsWe conducted a retrospective study of patients with clinical and biochemical characteristics compatible with GLUT1DS who were attended at the metabolic and neurodegenerative disease unit of a tertiary hospital between 2002 and 2018.
Genotypic dataA molecular genetic analysis of DNA from peripheral blood was performed at a reference laboratory. Samples were processed with PCR amplification and subsequently by sequencing of SLC2A1 exons and adjacent introns. Sequencing was performed with an ABI 3130 DNA sequencer, and electropherograms were compared against the reference sequence (ENST00000426263.9). Clinical exome sequencing was performed in the case of negative results for SLC2A1 mutations. The pathogenic variants identified were confirmed by Sanger sequencing of samples from the patient and his/her parents.
Phenotypic dataWe gathered data on epidemiological variables (sex, age) and first- and second-degree family history of symptoms compatible with GLUT1DS. We also collected data on disease onset and progression, which enabled the identification of the clinical phenotype in each patient. We observed 2 distinct phenotypes, the classic phenotype and an atypical phenotype; both phenotypes were further subclassified. The classic phenotype (CP) was defined by presence of refractory epilepsy and psychomotor delay. Two subtypes were established according to seizure onset: early onset, when seizures appeared before the age of 2 years (CP1), and late onset, when seizures appeared after 2 years of age (CP2). We also specified whether the patient presented CMD, paroxysmal movement disorder, or acquired microcephaly. Atypical phenotypes were further classified into 4 subgroups according to the most predominant symptom: CMD (AP1), with or without intellectual disability; exercise-induced paroxysmal dyskinesia (EIPD) (AP2), with or without epilepsy; early-onset absence epilepsy (AP3); or other (AP4). In the case of AP4, we specified whether the phenotype was potentially associated (epilepsy with myoclonic-atonic seizures, dystonic tremor, alternating hemiplegia, or other type of epilepsy or non-epileptic paroxysmal event).
We gathered data on relevant variables for determining the biochemical phenotype: CSF and serum glucose levels, CSF/serum glucose ratio, and CSF lactate concentration. We performed basic cytological and biochemical analyses of the CSF. Analyses were performed after a fasting period of at least 4–6hours (the time needed for CSF glucose levels to stabilise).3 Serum glucose levels were determined immediately before performing the lumbar puncture, as the procedure may cause stress hyperglycaemia.3 We used the cut-off values for brain glucose deficiency previously used for GLUT1DS6: in patients with compatible symptoms, values equal to or below the 10th percentile for glucose CSF levels, equal to or below the 25th percentile for the CSF/serum glucose ratio, and equal to or below the 90th percentile for CSF lactate concentration. Normal lactate concentration is a necessary condition for diagnosis of GLUT1DS, as it helps rule out such other causes of hypoglycorrhachia as bacterial or tuberculous meningitis and some inborn errors of metabolism.6
Other potentially useful complementary testsWe also collected data from FDG-PET studies, when available; findings consistent with previously reported imaging findings of the disease were considered suggestive of GLUT1DS.8
We also gathered data from neurophysiological studies performed before and after consumption of carbohydrate-rich foods; results were considered to indicate GLUT1DS in patients showing clear improvements after food intake, in terms of either quality of background activity or presence or severity of interictal abnormalities or epileptic seizures. These patients underwent video EEG monitoring.
Progression and response to ketogenic dietary therapiesWhen appropriate, we gathered data on the number of antiepileptic drugs (AED) received and their effectiveness in controlling seizures. We also recorded whether patients were on KDT, and the type and duration. We excluded patients showing poor adherence to the diet or not achieving sufficient levels of ketonaemia, for any reason. For the remaining patients, data were gathered on the general effectiveness of KDT and its effect on 3 variables: seizure control, control of motor alterations, and improvements in cognitive function (school performance or other). KDT was considered to be effective (or to achieve a significant improvement) when patients showed either complete or partial seizure control (>50% decrease in seizure frequency). Data were also gathered on the discontinuation of AEDs as a result of persistent seizure control. Regarding motor symptoms, in patients with episodic symptoms (EIPD or CMD with paroxysmal symptoms), complete resolution of these symptoms or a >50% decrease in the frequency of episodes was considered a significant improvement. In patients with permanent motor symptoms (mainly poor coordination, imbalance, and/or ataxia), KDT was considered effective when it achieved objective motor improvements (patients recovered the ability to walk or displayed an improvement in gait pattern, or performed better in daily living activities requiring coordination). Regarding cognitive function, we evaluated the acquisition of new skills (increased speed in performing a task, more complex tasks, etc.) following onset of KDT in children with moderate-to-severe intellectual disability. Improvements in attention, communication/interaction, and language were positively evaluated. Children with normal cognitive function or mild intellectual disability were considered to show adequate response to KDT when they displayed improvements in attention, reasoning, and school performance. These data were gathered from psychopaedagogical and/or school reports, neuropsychological tests, interviews with patients’ families, and clinical data gathered during follow-up consultations.
Regarding clinical progression, we gathered data on whether patients presented the following conditions during follow-up: cognitive impairment (and severity), microcephaly, movement disorders, and/or executive dysfunction. Cognitive function and alterations in specific functions were evaluated with neuropsychological tests and test batteries; the intelligence quotient was determined using the Wechsler Intelligence Scale for Children, fourth edition (WISC-IV) or the Wechsler Preschool and Primary Scale of Intelligence, fourth edition (WPPSI-IV), depending on the age. In patients unable to complete these tests (normally due to severe disease or inability to cooperate), the level of dysfunction was evaluated through clinical assessment at a specialised consultation with an expert paediatric neurologist and/or neuropsychologist.
Statistical analysisWe assessed the homogeneity of demographic variables, medical history, and other clinical parameters. Data are presented as mean, median, and standard deviation for quantitative variables, and absolute and relative frequencies for qualitative variables. Quantitative variables were analysed with the t test or the non-parametric Mann–Whitney U test, depending on whether they were normally distributed. Qualitative variables were analysed for homogeneity with the chi-square test or the Fisher exact test. Values of P<.05 were considered statistically significant. Statistical analysis was performed with the SPSS software (version 22.0; IBM Inc.).
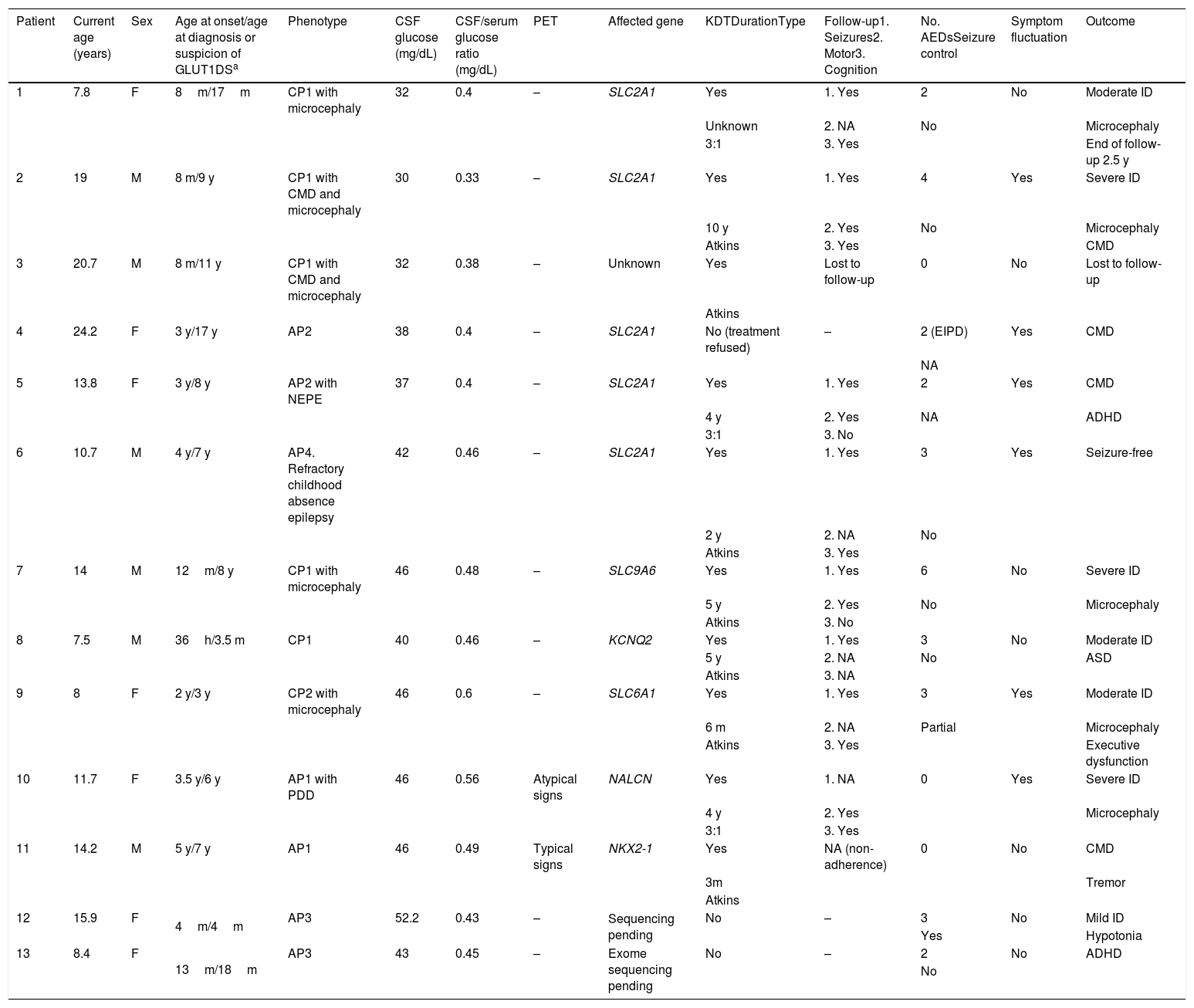
ResultsOur study included 13 patients (6 male, 7 female), who were followed up for 22 months to 14 years (Table 1). Only 2 patients had family history of GLUT1DS, and both showed positive genetic test results.
Clinical characteristics of our sample.
| Patient | Current age (years) | Sex | Age at onset/age at diagnosis or suspicion of GLUT1DSa | Phenotype | CSF glucose (mg/dL) | CSF/serum glucose ratio (mg/dL) | PET | Affected gene | KDTDurationType | Follow-up1. Seizures2. Motor3. Cognition | No. AEDsSeizure control | Symptom fluctuation | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 7.8 | F | 8m/17m | CP1 with microcephaly | 32 | 0.4 | – | SLC2A1 | Yes | 1. Yes | 2 | No | Moderate ID |
| Unknown | 2. NA | No | Microcephaly | ||||||||||
| 3:1 | 3. Yes | End of follow-up 2.5 y | |||||||||||
| 2 | 19 | M | 8 m/9 y | CP1 with CMD and microcephaly | 30 | 0.33 | – | SLC2A1 | Yes | 1. Yes | 4 | Yes | Severe ID |
| 10 y | 2. Yes | No | Microcephaly | ||||||||||
| Atkins | 3. Yes | CMD | |||||||||||
| 3 | 20.7 | M | 8 m/11 y | CP1 with CMD and microcephaly | 32 | 0.38 | – | Unknown | Yes | Lost to follow-up | 0 | No | Lost to follow-up |
| Atkins | |||||||||||||
| 4 | 24.2 | F | 3 y/17 y | AP2 | 38 | 0.4 | – | SLC2A1 | No (treatment refused) | – | 2 (EIPD) | Yes | CMD |
| NA | |||||||||||||
| 5 | 13.8 | F | 3 y/8 y | AP2 with NEPE | 37 | 0.4 | – | SLC2A1 | Yes | 1. Yes | 2 | Yes | CMD |
| 4 y | 2. Yes | NA | ADHD | ||||||||||
| 3:1 | 3. No | ||||||||||||
| 6 | 10.7 | M | 4 y/7 y | AP4. Refractory childhood absence epilepsy | 42 | 0.46 | – | SLC2A1 | Yes | 1. Yes | 3 | Yes | Seizure-free |
| 2 y | 2. NA | No | |||||||||||
| Atkins | 3. Yes | ||||||||||||
| 7 | 14 | M | 12m/8 y | CP1 with microcephaly | 46 | 0.48 | – | SLC9A6 | Yes | 1. Yes | 6 | No | Severe ID |
| 5 y | 2. Yes | No | Microcephaly | ||||||||||
| Atkins | 3. No | ||||||||||||
| 8 | 7.5 | M | 36h/3.5 m | CP1 | 40 | 0.46 | – | KCNQ2 | Yes | 1. Yes | 3 | No | Moderate ID |
| 5 y | 2. NA | No | ASD | ||||||||||
| Atkins | 3. NA | ||||||||||||
| 9 | 8 | F | 2 y/3 y | CP2 with microcephaly | 46 | 0.6 | – | SLC6A1 | Yes | 1. Yes | 3 | Yes | Moderate ID |
| 6 m | 2. NA | Partial | Microcephaly | ||||||||||
| Atkins | 3. Yes | Executive dysfunction | |||||||||||
| 10 | 11.7 | F | 3.5 y/6 y | AP1 with PDD | 46 | 0.56 | Atypical signs | NALCN | Yes | 1. NA | 0 | Yes | Severe ID |
| 4 y | 2. Yes | Microcephaly | |||||||||||
| 3:1 | 3. Yes | ||||||||||||
| 11 | 14.2 | M | 5 y/7 y | AP1 | 46 | 0.49 | Typical signs | NKX2-1 | Yes | NA (non-adherence) | 0 | No | CMD |
| 3m | Tremor | ||||||||||||
| Atkins | |||||||||||||
| 12 | 15.9 | F | 4m/4m | AP3 | 52.2 | 0.43 | – | Sequencing pending | No | – | 3 | No | Mild ID |
| Yes | Hypotonia | ||||||||||||
| 13 | 8.4 | F | 13m/18m | AP3 | 43 | 0.45 | – | Exome sequencing pending | No | – | 2 | No | ADHD |
| No |
ADHD: attention-deficit/hyperactivity disorder; AED: antiepileptic drug; AP: atypical phenotype; ASD: autistic spectrum disorder; CMD: complex movement disorder; CP: classic phenotype; CSF: cerebrospinal fluid; d: days; EIPD: exercise-induced paroxysmal dyskinesia; F: female; h: hours; ID: intellectual disability; KDT: ketogenic dietary therapy; m: months; M: male; NA: not applicable; NEPE: nonepileptic paroxysmal events; PDD: psychomotor developmental delay; PET: positron emission tomography; y: years.
The molecular genetic study of SLC2A1 detected pathological alterations in 5 patients. For data analysis, we included a sixth patient in this group, for whom molecular genetic study results were not available; this patient was diagnosed with GLUT1DS based on the detection of CSF glucose levels below 33mg/dL in 2 analyses, and the typical clinical phenotype. Additional genetic testing revealed pathogenic alterations in 5 of the remaining 7 patients; in 4 of these, the alterations affected genes encoding other channels or transporters.
Clinical and biochemical phenotypic data: comparative analysisMean age at symptom onset was one year (range, 0.01-5), with no differences between patients with and without mutations in SLC2A1 or between sexes. The classic phenotype was identified in 6 patients: 5 with early-onset epilepsy (CP1) and one patient with late-onset absence epilepsy and psychomotor developmental delay, presenting at the age of 2 years and 7 months (CP2). Patients with atypical phenotypes were distributed as follows: 2 presented CMD (AP1) (one also presented cognitive impairment), 2 had EIPD (AP2), 2 had early-onset absence epilepsy (AP3), and the remaining patient had refractory childhood absence epilepsy, which cannot be assigned to any of the other categories (AP4).
No significant differences were observed between patients with and without SLC2A1 mutations in terms of age at symptom onset (2.1 vs 1.9 years; P=.8), clinical form of presentation (classic phenotype, 50% vs 42.8%), presence of microcephaly (50% vs 42.8%), progression with intellectual disability, or severity of intellectual disability. All patients initially presenting psychomotor developmental delay (all patients with CP and one patient with AP1) presented moderate-to-severe intellectual disability during disease progression. The remaining patients, with initially normal psychomotor development, did not present cognitive impairment during follow-up. No statistically significant differences were observed in the presence of executive dysfunction (observed in 5 patients); executive function was assessed only in 7 patients, since the remaining patients had severe intellectual disability.
Patients with SLC2A1 mutations more frequently presented symptom fluctuations after food intake (66.7% vs 28.6%; P=.28) and persistence of motor symptoms during the follow-up period (66% vs 28.6%; P=.29).
These patients also presented significantly lower CSF glucose levels (34.5mg/dL [30–42] vs 46mg/dL [40–52]; P =.04) and CSF/serum glucose ratio (0.4 [0.3−0.46] vs 0.48 [0.43−0.6]; P=.05). No other significant findings were observed in the cytological or biochemical analyses.
Phenotypic data: descriptive analysis by groupAmong patients with SLC2A1 mutations, 3 presented the classic phenotype (patients 1, 2, and 3): all 3 presented early-onset epilepsy and microcephaly, and 2 also had CMD. These patients showed the lowest CSF glucose levels in the series (≤32mg/dL). The remaining 3 patients with SLC2A1 mutations (patients 4, 5, and 6) presented atypical phenotypes; 2 (patients 4 and 5) presented EIPD, with no associated epilepsy or cognitive impairment. Patient 6 presented refractory childhood absence epilepsy; a maternal uncle had presented similar symptoms, and the mutation was detected in his mother, who is asymptomatic. Patients with SLC2A1 mutations and atypical phenotypes presented symptoms at a later age than those with the classic phenotype, also displaying higher CSF glucose levels.
Three of the patients without SLC2A1 mutations and presenting other genetic alterations displayed the classic phenotype (patients 7, 8, and 9). Patient 7 presented early-onset epilepsy (at 12 months of age), psychomotor developmental delay, and microcephaly; epilepsy, but not cognitive function, improved significantly with KDT. Massive sequencing identified a presumably severe novel hemizygous mutation in the SLC9A6 gene (c.803+1G>A), which encodes sodium-proton exchanger SLC9A6; alterations in the gene may cause Christianson syndrome,14 an X-linked disorder (Xq26.3) characterised by severe intellectual disability, absent language development, autistic spectrum disorder, epilepsy, microcephaly, late-onset ataxia, muscle weakness, and dystonia. A genetic study detected the same mutation in his mother. Patient 8 presented neonatal epilepsy, which was refractory to combination therapy and showed immediate, complete response to KDT (the patient is currently seizure-free after 6 years receiving this treatment). He later developed autistic spectrum disorder. Genetic studies detected a heterozygous allelic variant previously reported as pathogenic (c.619C>T, p.Arg207Trp) in the KCNQ2 gene (20q13.33), described in patients with benign neonatal epilepsy and more recently in patients with different types of early-onset epileptic encephalopathies.15 Patient 9 presented epilepsy at the age of 2 years and 7 months, with episodes of vertical gaze deviation and initially no apparent disconnection from her surroundings. She later developed absence seizures, which were detected during fasting video EEG; seizures resolved approximately 30minutes after consumption of sugars, and epileptiform abnormalities also decreased considerably. A de novo pathogenic variant (c.T277delGC, p.Ala93Glyfs*113) was identified in the SLC6A1 gene (3p25.3). The variant had not been described previously in the genetic databases consulted, and is likely to be pathogenic since it results in protein truncation. The SLC6A1 gene encodes a gamma-aminobutyric acid transporter located in the cell membrane; mutations in this gene have been associated with myoclonic atonic epilepsy, also known as Doose syndrome.16
The remaining 2 patients with mutations in SLC2A1 but presenting other genetic alterations (patients 10 and 11) presented atypical phenotypes. Patient 10 had history of psychomotor developmental delay from early childhood, and subsequently presented episodes of neck and upper limb dystonia progressing to CMD with ataxia, dystonia, and tremor. She also presented episodes of hypoactivity that improved with food intake. KDT (initial ratio 3:1) significantly improved dystonia, motor disorder, and cognitive function; improvements persist after 5 years with a low glycaemic diet. This patient displayed a de novo mutation (p.Ile322Thr) in the NALCN gene (13q32.3-q33.1), which encodes an ion channel; mutations in this gene are associated with CLIFAHDD syndrome (congenital contractures of the limbs and face, hypotonia, and developmental delay).17,18 Patient 11 presented poor coordination and gait alterations, which worsened at 3 years of age; he subsequently developed CMD, characterised by dystonia and tremor, and presented no associated cognitive impairment. He was treated with levothyroxine from onset. A dystonia panel yielded negative results, and an FDG-PET study revealed signs compatible with GLUT1DS. Massive sequencing identified a novel heterozygous allelic variant (c.727delC, p.Arg243Alafs*4) in the NKX2-1 gene (14q33.3), which is not included in the professional version of the Human Gene Mutation Database or in population databases; bioinformatic prediction tools indicated pathogenicity. NKX2-1 seems to be involved in the development of the prosencephalon, thyroid glands, and lungs during embryonic development; alterations in the gene have been associated with choreoathetosis and congenital hypothyroidism with or without pulmonary dysfunction, a syndrome following an autosomal dominant inheritance pattern.19
Complementary test resultsOnly 2 patients (patients 10 and 11) underwent FDG-PET studies (neither showed SLC2A1 mutations), one of whom displayed signs compatible with GLUT1DS (patient 11).
Response to ketogenic dietary therapiesAll patients with SLC2A1 mutations and epileptic seizures were refractory to AED combination therapy, whereas 3 of the 7 patients without SLC2A1 mutations showed adequate response to AED, although seizures were initially poorly controlled.
Ten patients started KDT, lasting between 6 months and 8 years and 10 months (median, 4.5 years); 8 of these (4 with SLC2A1 mutations) showed good adherence and achieved ketosis. All patients with SLC2A1 mutations showed significant improvements with this approach. Three (patients 1, 2, and 6) presented frequent episodes (daily or monthly) of refractory seizures, and the remaining patient (patient 5) had EIPD and nonepileptic paroxysmal events of uncertain aetiology (symptoms were poorly compatible), associated with bursts of generalised paroxysmal activity in EEG. Patients 1 and 6 showed complete response to KDT, remaining seizure-free from the onset of treatment. Patient 2 improved considerably, presenting seizures only occasionally. In all 3 patients, these marked improvements enabled progressive discontinuation of antiepileptic treatment, from combination therapy with 2-3 AEDs to nearly complete discontinuation. However, seizures reappeared after discontinuation of the last AED, which had to be resumed; patients finally achieved seizure control with KDT plus one AED. All of them showed improvements in attention and/or communication. In patient 5, paroxysmal events (both those associated with EIPD and nonepileptic paroxysmal events) and paroxysmal EEG activity resolved after onset of KDT. Throughout progression, she has presented isolated episodes of paroxysmal dyskinesia, always in the context of high-intensity physical exercise. In the group of patients without SLC2A1 mutations, improvements were observed in the 4 patients showing mutations in genes encoding other channels. The only patient without mutations in these genes discontinued KDT 3 months after onset due to lack of effectiveness; however, it should be noted that adherence was suboptimal. Two of the patients showing improvements (patients 8 and 9) presented refractory epilepsy with multiple daily episodes; in both cases, seizures disappeared with KDT plus AED treatment. The remaining patient (patient 10) presented episodes of dystonia, occasionally associated with rigidity of one side of the body; the frequency of these symptoms decreased progressively until complete resolution. Patients 9 and 10 also displayed mild improvements in attention and cognitive function.
Age of onset of KDT varied greatly, ranging from 4 months (patient 8) to 9.5 years (patient 2); age of onset was not found to be exclusively responsible for patient response to the ketogenic diet.
DiscussionThe increasing complexity and variability of GLUT1DS phenotypes and genotypes and the lack of a clear genotype-phenotype correlation represent a challenge in the diagnosis and treatment of patients presenting clinical and biochemical findings compatible with the syndrome. Our results reflect this complexity: patients with SLC2A1 mutations presented great phenotypic variability, and their phenotypes overlapped with those of patients without SLC2A1 mutations to the point of behaving as phenocopies.2,9 Our results should be interpreted with caution given the small size of our sample; however, our sample may be more representative of clinical practice. Thus, the previously reported overlap and difficulty distinguishing between patients with and without SLC2A1 mutations2,9 are also observed in our comparison of clinical characteristics: no significant differences were observed between groups in the frequency of the classic or atypical phenotype, microcephaly, or intellectual disability (or its severity).
A non-significant difference was observed in the prevalence of movement disorders (whether at onset or during disease progression), with a higher frequency among patients with SLC2A1 mutations; furthermore, EIPD was only observed in these patients. This characteristic has been well documented in patients with GLUT1DS, and constitutes an increasingly recognised form of presentation of the syndrome, especially forms presenting without epilepsy and with normal or mildly impaired cognitive function.2–4,19 Increased detection both of this syndrome and of other non-epileptic movement disorders (which are frequently considered less severe forms of the disease) has sparked interest in applying KDT to this type of symptoms, which are presumably less severe than epilepsy but are nonetheless disabling.2,4,20 In our study, KDT was indicated for all patients with clinical phenotypes including CMD or EIPD, and achieved motor improvements in all patients showing adequate adherence (monitored with routine testing for ketosis) for a sufficient time. We were surprised to observe the positive response of the patient with CMD and no SLC2A1 mutations but presenting a de novo mutation in the NALCN gene (mutations in this gene cause CLIFAHDD syndrome). Symptom fluctuation or worsening under fasting conditions was more frequent in patients with SLC2A1 mutations than in those with other mutations, although the difference was not statistically significant. However, 2 patients without SLC2A1 mutations did display symptom fluctuations: one had a NALCN mutation and the other had a SLC6A1 mutation (the latter displayed improvements in video-EEG recordings after food intake).
Regarding the overall effectiveness of KDT (excluding patients with poor adherence or following the diet for insufficient time), all patients with SLC2A1 mutations showed clinical improvements. In the group of patients without these mutations, KDT also improved symptoms in all patients with mutations in genes encoding other ion channels. No predictive parameters for response to KDT are currently available.2,12,13 According to previous research, the degree of response to KDT even varies between individuals with the same SLC2A1 mutation.12,13 Presence of a mutation in the SLC2A1 gene seems not to be the only factor determining response to KDT; in fact, several studies including patients without SLC2A1 mutations suggest the involvement of post-transcriptional factors in the encoded protein and even of other modifier genes and proteins in the pathophysiology of this complex entity. This type of association may mediate good response to KDT and the changes in video-EEG activity occurring after food intake in our patients without SLC2A1 mutations but with mutations in genes encoding other channels; thorough biochemical studies would be necessary to confirm this hypothesis.
In the light of the above, GLUT1DS should be considered in patients with any of the compatible phenotypes described. This may enable early diagnosis and indication of KDT, which in many cases achieves marked clinical improvements and even complete resolution of some symptoms, regardless of whether subsequent genetic studies detect a mutation in SLC2A1. KDT enables the discontinuation of AED treatment or a reduction in the number of AEDs necessary to control seizures, and may even help prevent the indication of more invasive approaches. Only biochemical parameters displayed statistically significant differences between groups, with CSF glucose levels and CSF/serum glucose ratio being significantly lower in patients with SLC2A1 mutations. Patients with EIPD and normal cognitive function presented higher values, as reported in previous studies,4,6,10 although there may be an overlap with patients without SLC2A1 mutations.
ConclusionsThe increasing complexity and variability of GLUT1DS phenotypes and genotypes and the lack of a clear genotype-phenotype correlation represent a challenge in the diagnosis and treatment of patients presenting clinical and biochemical findings compatible with the syndrome, particularly in those without SLC2A1 mutations.
Funding“Identification and clinical and biochemical characterisation of patients with GLUT1DS: treatment monitoring.” Translational research project 2017, CIBERER. Coordinator: Dr Luis González Gutiérrez-Solana (GCV6). Participating units: U703 (Artuch); U746 (Pérez); GCV5 (Couce); GCV6 (Gutiérrez-Solana); GCV7 (López Laso); GCV8 (del Toro). Research project: hereditary metabolic disorders.
Conflicts of interestAll authors approved the content of the manuscript and have no conflicts of interest to declare.
Please cite this article as: Jiménez Legido M, Cortés Ledesma C, Bernardino Cuesta B, López Marín L, Cantarín Extremera V, Pérez-Cerdá C, et al. Estudio de pacientes pediátricos con fenotipo clínico y bioquímico de síndrome de déficit de transportador de glucosa cerebral (GLUT-1). Neurología. 2022;37:91–100.
