





Los dispositivos intrauterinos hormonales de levonorgestrel son métodos anticonceptivos seguros y eficaces. Al igual que los medicamentos convencionales, una vez expirada la patente, las autoridades regulatorias de salud pueden aprobar el registro de productos similares. El objetivo de ello es disminuir los costos, considerando el elevado precio del producto original. Este tipo de productos están regulados y se aplican requisitos generales similares a los medicamentos tradicionales para demostración de seguridad y eficacia. Las propiedades mecánicas únicas del producto de referencia plantean un gran desafío a los productos similares. El presente artículo analiza de manera comparativa las características de los diversos sistemas intrauterinos hormonales de levonorgestrel, disponibles en el mercado. La autoridad sanitaria y los diversos centros clínicos deben considerar que en este tipo de productos no hay, hasta la fecha en el mundo, genéricos intercambiables y que por lo tanto, se debe decidir la intercambiabilidad de éstos sobre la base de estudios de bioequivalencia in vivo, luego de la demostración de equivalencia farmacéutica in vitro, tal y como sugiere la FDA, o en su defecto deberían ser registrados como productos nuevos, con estudios clínicos apropiados que demuestren seguridad y eficacia.
Levonorgestrel hormonal intrauterine systems are safe and effective contraceptive methods. Like conventional drugs, once the patent expires, health regulatory authorities can approve the registration of similar products. The objective of this is to reduce costs, considering the high price of the original product. These types of products are regulated as drugs and similar general requirements apply to traditional drugs for demonstration of safety and efficacy. The unique mechanical properties of the reference product pose a great challenge to similar products. This article comparatively analyzes the characteristics of the various levonorgestrel hormonal intrauterine systems available on the market. Therefore, the health authority and clinical centers must consider that up to date, there are no interchangeable generics in this type of products worldwide. Thus, their interchangeability must be decided on the basis of in vivo bioequivalence studies after the demonstration of in vitro pharmaceutical equivalence, as suggested by the FDA. Without that, they should be registered as new products, with appropriate clinical studies that demonstrate safety and efficacy.
La regulación de fertilidad beneficia a las personas y a la población, ya que protege el derecho a la vida y la salud, apoya el derecho de mujeres y hombres a disfrutar de su sexualidad y reproducción y el derecho de los hijos e hijas a nacer siendo deseados. Además, resguarda la libertad de conciencia de las personas para decidir utilizar o no algún método anticonceptivo, a partir de sus valores personales, resguarda el principio de no-maleficencia y el principio de equidad y justicia. Esto se cumple cuando los servicios son accesibles a todas las personas sin discriminación y cuando las autoridades de estos servicios se responsabilizan de facilitar y supervisar que esta condición se respete1.
La anticoncepción emplea medicamentos, dispositivos o cirugía para prevenir el embarazo. A todos estos medios se les conoce como métodos anticonceptivos2. En la actualidad, se tiene disponibilidad de diferentes tipos de métodos anticonceptivos, los cuales se clasifican en las siguientes categorías: métodos hormonales, métodos de barrera, anticonceptivos reversibles de larga duración, anticonceptivos de emergencia y esterilización1. Todos ellos deben cumplir con las condiciones básicas de eficacia, seguridad, aceptabilidad, disponibilidad y reversibilidad1.
Dentro de los métodos actuales para este fin, están los dispositivos intrauterinos (DIUs), que son un método de anticoncepción seguro y altamente efectivo. Hasta el momento se encuentran disponibles en el mercado dos tipos generales de DIUs: la T con Cobre (99,2% de efectividad) y los DIU que liberan levonorgestrel (LNG) (99,8% de efectividad)3. De este último tipo, Mirena® 52mg LNG, inventado por Tapani Luukkainen (Finlandia) y desarrollado por el Population Council a fines de la década de 1980 y más tarde por Schering Oy y Schering AG, fue aprobado en 1993 en Finlandia y en varios países más al mismo tiempo4, fue el primero en ser aprobado por la FDA5 para su uso y comercialización en Estados Unidos, en el año 2000, y es considerado el “Gold Standard” de este tipo de DIU4. Posteriormente, otros DIU-LNG han salido al mercado tras su aprobación por las entidades regulatorias.
En Estados Unidos, los DIU-LNG están inscritos como nuevas aplicaciones de medicamentos (NDA-New Drug Applications)6. Hasta la fecha, en Chile, este tipo de anticonceptivos se encuentra bajo la categoría de fármaco7 y no se solicitan pruebas de equivalencia terapéutica o bioequivalencia para la inscripción o registro de copias, a pesar de que la FDA sí lo recomienda8.
La bioequivalencia es una condición que permite establecer equivalencia terapéutica de un producto similar mediante un estudio farmacocinético de biodisponibilidad comparativa (o bio-exenciones, cuando apliquen), por lo tanto, respalda la misma eficacia y seguridad que el producto comparador o innovador. Esta condición hace que un producto equivalente terapéutico sea intercambiable, cuando además sea equivalente farmacéutico, tenga un registro sanitario actualizado y su proceso de manufactura validado. Estos estudios pueden ser in vivo, en voluntarios sanos para establecer biodisponibilidad homóloga, in vitro mediante una prueba que se acepta extrapola la situación al interior del organismo (ej. perfiles de disolución, ensayos de permeabilidad intestinal, etc.) o ex vivo, extrayendo tejido o muestras de individuos para evaluar los niveles locales del medicamento9–11.
De acuerdo con lo anteriormente señalado, el objetivo de este manuscrito es revisar los antecedentes generales de los DIU-LNG y analizar desde el punto de vista técnico-científico la necesidad del requerimiento de ensayos de bioequivalencia in vivo, ex vivo o in vitro para los DIU-LNG en el procedimiento de registro como copias, similares o fármacos multi-fuente, con respecto al producto original o innovador.
DIU de levonorgestrelEl DIU-LNG es un dispositivo plástico de polietileno en forma de T (332mm). Un manguito de elastómero está montado alrededor de su parte vertical. Esta manga consta de una mezcla 1 a 1 de polidimetilsiloxano (PDMS) y LNG liberado. La manga está cubierta con una membrana de control de liberación de fármaco de PDMS de grado médico y un hilo de recuperación para retirar el dispositivo, según sea necesario (Figura 1)12. La hormona liberada (LNG) hace que el moco cervical se engrose, inhibe la llegada del esperma al óvulo y su fecundación, afina el revestimiento uterino y puede impedir la ovulación13,14. La tasa de falla del DIU hormonal es inferior a 0,5% (menos de 1 de cada 200 mujeres que lo usan quedan embarazadas en 1 año de uso); sin embargo, aproximadamente el 4,5% de las mujeres pueden experimentar la expulsión del dispositivo15 y un profesional tendrá que volver a insertar otro, uno nuevo, excepto en caso de perforación uterina o si ocurre un dolor excepcional o sangrado después de la inserción16.

Sistema/dispositivo intrauterino de administración de progestina 12.
Este método también se puede usar para tratar la menorragia, dado que la hormona reduce el sangrado15. Las biopsias del endometrio con el DIU-LNG colocado exhiben atrofia glandular y estroma decidualizado y los resultados no difieren según el tiempo que lleva colocado el DIU o el tipo de DIU-LNG en cuanto a su potencia17. La tasa de liberación de fármaco de estos productos DIU-LNG disminuye gradualmente con el tiempo18. Este sistema libera LNG durante un período prolongado de hasta 5 años a una tasa prácticamente constante, aunque es más alta al inicio de la colocación del producto y decae hasta el final de su uso en general aproximadamente entre un 50-60%19. Con la administración local, las concentraciones de LNG son altas en el tejido endometrial, pero no lo son en los tejidos adyacentes, incluidos el miometrio y las trompas de Falopio. Si bien las concentraciones de LNG en el miometrio y las trompas de Falopio se encuentran en el mismo rango que aquellas obtenidas mediante la administración oral de 30μg de LNG, las concentraciones endometriales son 200 a 800 veces más altas con el DIU- LNG18.
Los estudios clínicos del producto han informado que los efectos adversos más frecuentemente reportados (>10%) son: cefalea (≤16%), migraña (≤16%), acné vulgar (6% a 15%), seborrea (1% a 15%), menorragia (5% a 59%; generalmente el sangrado disminuye después de los primeros 90 días), amenorrea (≤42%, aumenta con la duración del tratamiento), metrorragia (23%), quiste ovárico (5% a 22%; incluye quistes sintomáticos y asintomáticos), dolor abdominal (≤23%) sangrado uterino anormal (52%), menstruaciones irregulares (25% a 43%; la menstruación se vuelve más regular después de los primeros 90 días), vulvo-vaginitis (11% a 24%), dolor pélvico (≤23%), infección vaginal (9% a 19%), infección vulvo-vaginal (8% a 19%), flujo vaginal (4% a 15%)16.
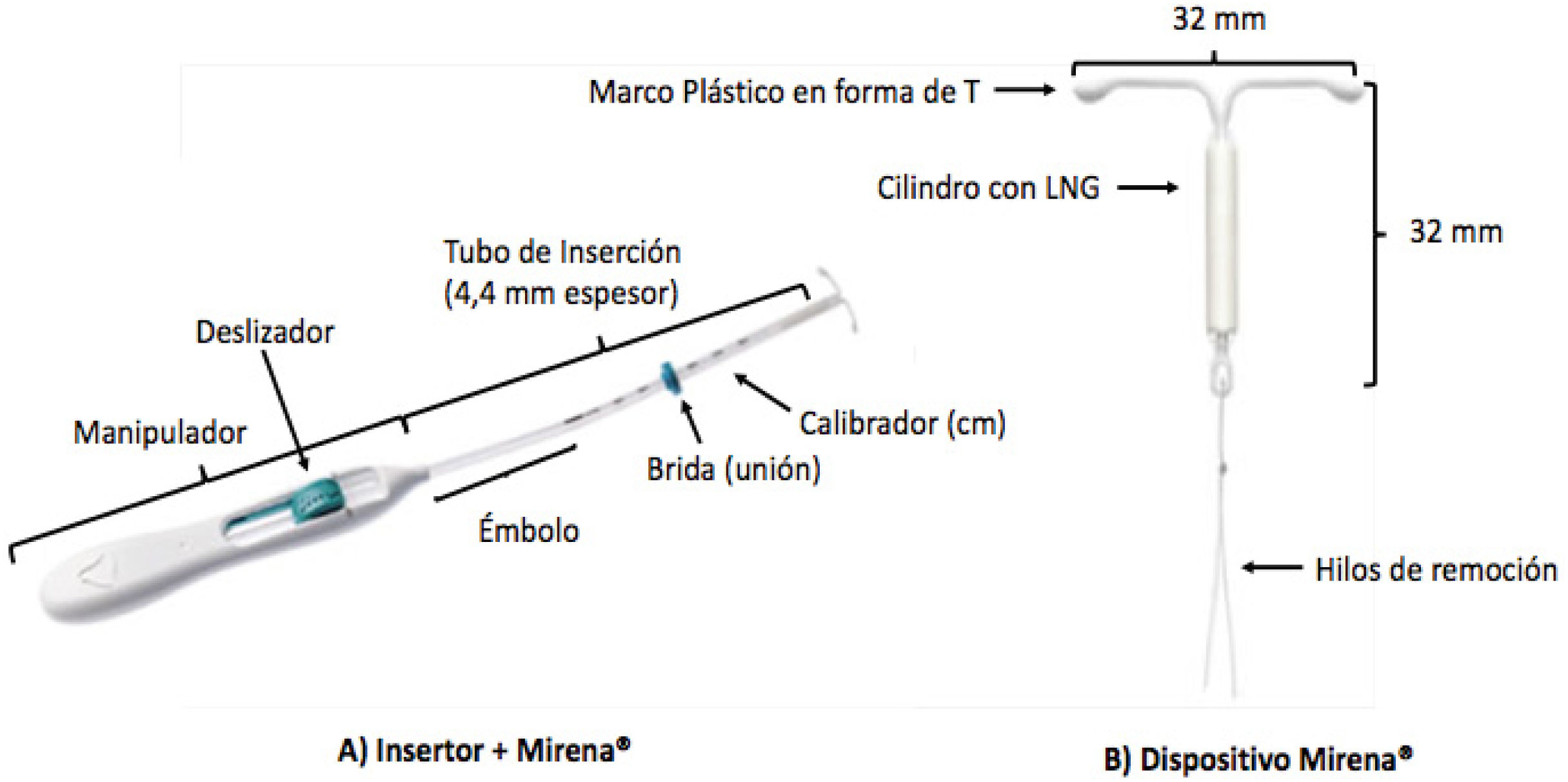
El primer DIU-LNG conocido como Mirena® comercializado por Bayer Healthcare, fue desarrollado en el año 1980 y recibió la aprobación de la FDA en el año 2000 para comercialización en EE.UU., posee un almacenamiento de 52mg de LNG con una tasa de liberación de 20μg/24hrs. Dentro de sus excipientes presenta elastómero de PMDS, sílice coloidal anhidra, polietileno, sulfato de bario y óxido de hierro20,21. El brazo vertical está cargado en el tubo de inserción en la punta de un insertor o insertador. El DIU y el insertador están esencialmente exentos de impurezas visibles (Figura 2). Este dispositivo fue patentado en 1999 (n° patente US6476079 (B1), fecha 23/12/1999) y tiene actualmente tres patentes vigentes (US10,561,52422; US9,615,96523 y US9,668,912)24, que protegen este medicamento junto al insertador. Este producto tiene en total 45 patentes asociadas, en veinte países.
. B. Dispositivo solo. LNG: Levonorgestrel. (adaptado de Ref. 20).")
Además, existen 3 NDAs (New Drug Applications) que establecen la aprobación de Mirena® el 06/12/2000 y de sus productos homólogos con dosificaciones más bajas, Skyla® y Kyleena®, con fechas de expiración 16/09/2029 y 01/04/20315,25,26.
También existe una exclusividad relacionada a la comercialización otorgada por la FDA a este DIU-LNG tras su aprobación, hasta el 20/08/2023, para la indicación “Régimen de dosificación que extiende el uso de anticonceptivos de 5 años hasta 6 años”26.
En Chile por su parte, se registra la patente nacional n° CL48227 concedida el 17/02/2012, que corresponde a la patente de Mirena®. Se registran además dos solicitudes de patente para el sistema con insertador, la solicitud n° 201900929 de fecha 05/04/2019 y la solicitud N° 201900977 de fecha 11/04/2019, ninguna de ellas tiene aún resolución. El producto similar, Lilleta® de la empresa Medicines360, por su parte, está protegido en la patente nacional CL56908, concedida con fecha 28/01/201927.
Por otro lado, en el sistema de registros nacionales del Instituto de Salud Pública de Chile (ISP) aparecen los siguientes registros28:
- •
F-4298/20: Mirena® (Bayer S.A.), sistema intrauterino 52mg (LNG), registrado el 28/06/2000.
- •
F-19985/18: Jaydess® (Bayer S.A.), sistema intrauterino 13,5mg (LNG micronizado), registrado el 03/06/2013.
- •
F-23076/16: Kyleena® (Bayer S.A.), sistema intrauterino 19,5mg (LNG), registrado el 17/09/2016.
- •
F-23975/18: Asertia® (Recalcine®), sistema intrauterino 20μg/24hrs, registrado el 04/09/2018.
- •
F-25974/21 y F-25993/21: Levosert® (Gedeon Richter®), sistemas intrauterinos 20μg/24hrs. en dos presentaciones, registradas el 10 febrero de 2021 y el 22 de febrero de 2021, respectivamente.
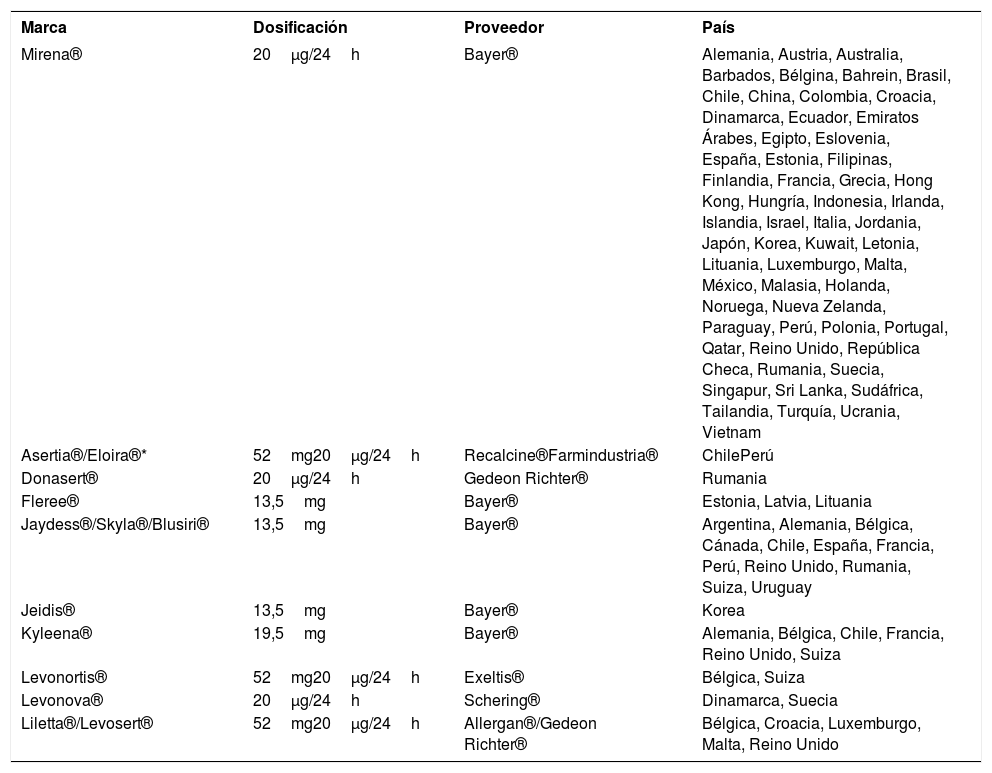
Hasta la fecha no hay productos genéricos de Mirena®. Sin embargo, otros DIU-LNG alternativos de buena calidad han salido al mercado tras su aprobación por FDA y otras entidades regulatorias (Tabla 1).
Dispositivos de administración intrauterina de levonorgestrel
| Marca | Dosificación | Proveedor | País |
|---|---|---|---|
| Mirena® | 20μg/24h | Bayer® | Alemania, Austria, Australia, Barbados, Bélgina, Bahrein, Brasil, Chile, China, Colombia, Croacia, Dinamarca, Ecuador, Emiratos Árabes, Egipto, Eslovenia, España, Estonia, Filipinas, Finlandia, Francia, Grecia, Hong Kong, Hungría, Indonesia, Irlanda, Islandia, Israel, Italia, Jordania, Japón, Korea, Kuwait, Letonia, Lituania, Luxemburgo, Malta, México, Malasia, Holanda, Noruega, Nueva Zelanda, Paraguay, Perú, Polonia, Portugal, Qatar, Reino Unido, República Checa, Rumania, Suecia, Singapur, Sri Lanka, Sudáfrica, Tailandia, Turquía, Ucrania, Vietnam |
| Asertia®/Eloira®* | 52mg20μg/24h | Recalcine®Farmindustria® | ChilePerú |
| Donasert® | 20μg/24h | Gedeon Richter® | Rumania |
| Fleree® | 13,5mg | Bayer® | Estonia, Latvia, Lituania |
| Jaydess®/Skyla®/Blusiri® | 13,5mg | Bayer® | Argentina, Alemania, Bélgica, Cánada, Chile, España, Francia, Perú, Reino Unido, Rumania, Suiza, Uruguay |
| Jeidis® | 13,5mg | Bayer® | Korea |
| Kyleena® | 19,5mg | Bayer® | Alemania, Bélgica, Chile, Francia, Reino Unido, Suiza |
| Levonortis® | 52mg20μg/24h | Exeltis® | Bélgica, Suiza |
| Levonova® | 20μg/24h | Schering® | Dinamarca, Suecia |
| Liletta®/Levosert® | 52mg20μg/24h | Allergan®/Gedeon Richter® | Bélgica, Croacia, Luxemburgo, Malta, Reino Unido |
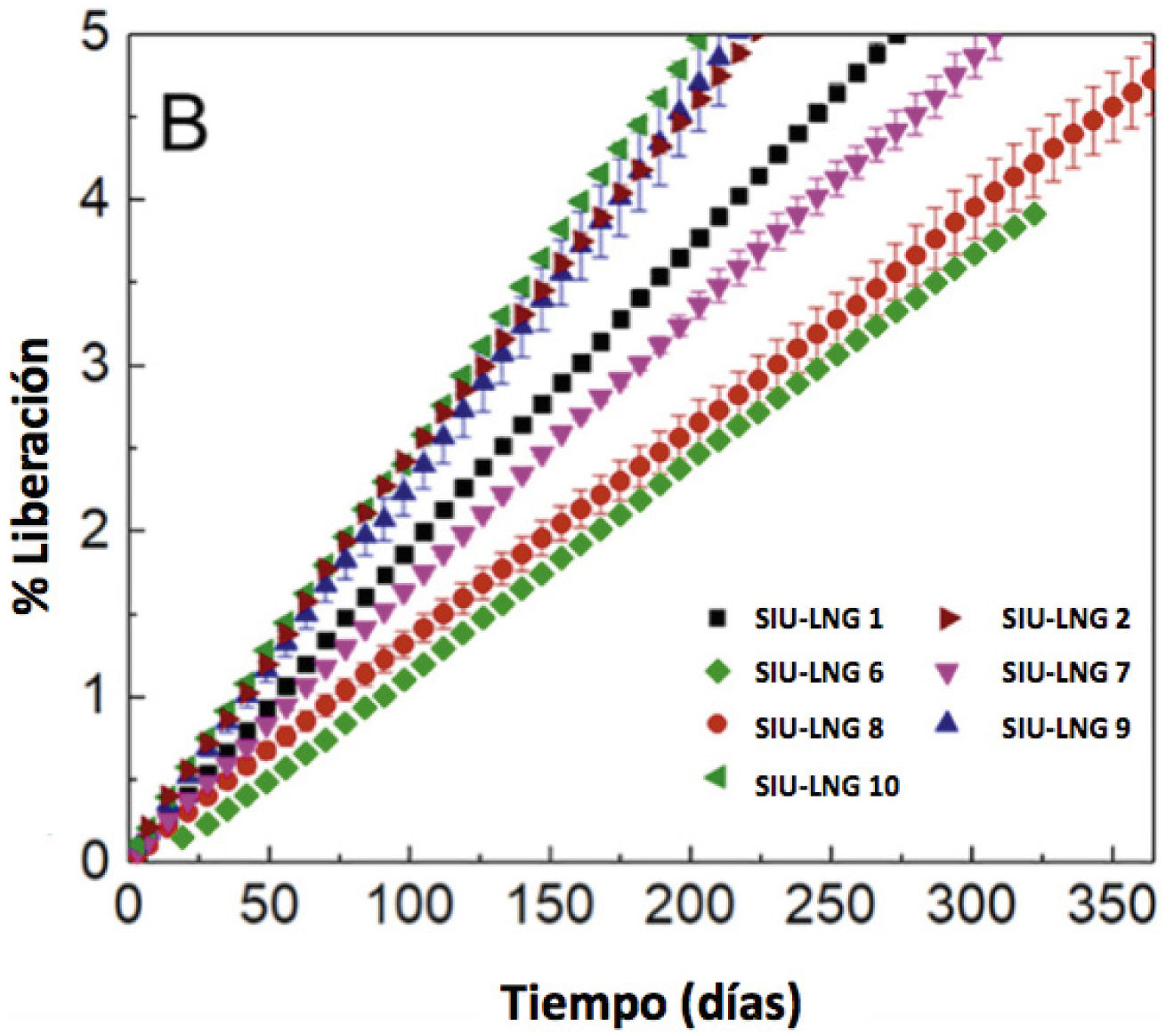
Estudios realizados por Bao y sus colaboradores19,29 han demostrado que, a pesar de la similitud entre algunos DIU-LNG, las tasas de liberación diaria y la dosis a 1 año y a 5 años varían para cada producto. Este tipo de fluctuaciones también se observa para productos de la misma configuración mecánica, pero de menor dosificación tales como Kyleena® y Jaydess® (Tabla 2)19,31. Del mismo modo Bao y cols.30 han demostrado que configuraciones de DIU-LNG que varían en el diámetro interno del reservorio, el origen de la materia prima de LNG y el origen de la membrana externa poseen diferente perfil de liberación de LNG (Tabla 3, Figura 3).
Tasa de liberación in vivo de los DIU-LNG aprobados por FDA para su uso en EE.UU
| Producto y cantidad total de LNG | Año de aprobación por FDA | Liberación diaria in vivo (μg) | Dosis antes de 1 año (μg/d) | Dosis al cabo de 1 año (μg/d) | Al final de los años aprobados para su uso (μg/d) | Promedio (μg/d) |
|---|---|---|---|---|---|---|
| Mirena®52 mg | 2000 | 20,0 | 20,0 (3 meses) | 18,0 | 10,0 (5 años) | 17,1 |
| Kyleena®19,5 mg | 2013 | 9,0 | 17,5 (24 días) | 9,8 | 7,4 (5 años) | 9,0 |
| Jaydess®13,5 mg | 2015 | 8,0 | 14,0 (24 días) | 6,0 | 5,0 (3 años) | 6,0 |
| Liletta®52 mg | 2016 | 8,0 | 19,5 (inicial) | 17,0 | 9,8 (5 años) | 14,1 |
Datos obtenidos desde Bao y cols, 2019 y OSHU, 201919,31.
Parámetros de diseño del producto para manufacturar DIU-LNG
| Formulación | *Diámetro interno de reservorio (mm) | Fuente del principio activo | Fuente de la membrana externa | Comentarios |
|---|---|---|---|---|
| DIU 1 | 1,5 | T | A (D: 2,41 mm) | Referencia |
| DIU 2 | 1,5 | T | B (D: 2,41 mm) | Variación de la fuente de la membrana externa |
| DIU 6 | 1,5 | T | A (D: 3,18 mm) | Membrana externa con una pared más gruesa |
| DIU 7 | 1,5 | C | A (D: 2,41 mm) | Mayor tamaño de partícula de la droga |
| DIU 8 | 0 | T | A (D: 2,41 mm) | Depósito no hueco |
| DIU 9 | 2 | T | A (D: 2,41 mm) | Mayor diámetro interno del reservorio |
| DIU 10 | 1,5 | T | A (D: 2,41 mm) | Reservorio de dos segmentos |
DIU: Dispositivo intrauterino, DE: diámetro exterior; T: Principio activo de Tecoland Corporation; C: Principio activo de Cayman Chemical; A: Dow Corning Silastic® (tubos de laboratorio); B: NuSil (grado farmacéutico/biomédico); D: diámetro.
 de LNG a partir del preparado equivalente cualitativa y cuantitativamente en diferentes DIU Condiciones Cloruro de sodio 0,9% p/v a 37°C en baño de agua con agitación a 100rpm, n=3. B. Ampliación de la vista del perfil de liberación en el rango de 0-5% de droga liberado durante el primer año. Adaptado de Ref. 30.")
A. Perfiles de liberación en tiempo real (1 año) de LNG a partir del preparado equivalente cualitativa y cuantitativamente en diferentes DIU
Condiciones Cloruro de sodio 0,9% p/v a 37°C en baño de agua con agitación a 100rpm, n=3. B. Ampliación de la vista del perfil de liberación en el rango de 0-5% de droga liberado durante el primer año. Adaptado de Ref. 30.
Las agencias reguladoras de medicamentos mundiales tienen requisitos para la aprobación de anticonceptivos que exceden los requisitos para otros medicamentos. Estos requisitos adicionales existen porque: 1) la prevención de embarazos no deseados no se considera una terapia curativa o profilaxis; 2) las receptoras de medicamentos anticonceptivos todavía están expuestas al riesgo de embarazo porque ningún anticonceptivo es 100% efectivo; y 3) los medicamentos anticonceptivos se toman durante períodos más prolongados que la mayoría de los demás medicamentos. Sin embargo, los estándares regulatorios para medicamentos y dispositivos médicos varían entre países, los que probablemente estén relacionadas con las diferencias internacionales en los esfuerzos de desarrollo de anticonceptivos, los factores sociales y económicos de cada país32.
Los anticonceptivos como los DIU, los diafragmas y los condones están regulados como dispositivos médicos por la FDA33. Aunque las formas reglamentarias y la terminología aplicables a los dispositivos difieren de las aplicables a los medicamentos, y los problemas específicos de seguridad varían entre los medicamentos y los dispositivos, requisitos generales sustancialmente similares para la demostración de la seguridad y la eficacia y una relación favorable de beneficios a riesgos se aplican a ambas categorías de productos, este es el caso de los DIU-LNG8.
Para LNG-DIU, la FDA recomienda estudios de bioequivalencia tanto in vitro como in vivo/ex vivo8. Además indica que para ser elegible para los estudios de bioequivalencia el producto de prueba debe cumplir con los siguientes criterios: i) cualitativamente y cuantitativamente igual que el medicamento de referencia ii) características fisicoquímicas y mecánicas equivalentes que incluyen: 1) tamaño de partícula y distribución del tamaño del ingrediente activo, 2) grado de reticulación del elastómero PDMS utilizado en el depósito de fármaco y en la membrana que controla la tasa de liberación del fármaco, 3) propiedades mecánicas del depósito de fármaco y de la membrana que controla la velocidad de liberación del fármaco, 4) apariencia, memoria, propiedades mecánicas del cuerpo en T y 5) fuerza de rotura del hilo de extracción comparable al estándar de referencia. Mismas dimensiones con respecto a cada componente que el estándar de referencia8.
En Chile los DIU están clasificados como dispositivos médicos. La definición de dispositivo médico indica que son: “cualquier instrumento, aparato, aplicación, material o artículo, incluyendo software, usados solos o en combinación y definidos por el fabricante para ser usados directamente en seres humanos, siempre que su acción principal prevista en el cuerpo humano no se alcance por medios farmacológicos, inmunológicos o metabólicos, aunque puedan concurrir tales medios a su función; con el propósito de diagnóstico, prevención, seguimiento, tratamiento o alivio de una enfermedad, daño o discapacidad; de investigación o de reemplazo o modificación de la anatomía o de un proceso fisiológico, o de regulación de la concepción” (Reglamento N° 825/98, Art. 2°, N°1)34. Haciendo referencia a que algún medio químico puede concurrir en la acción del dispositivo. Sin embargo, los DIU-LNG se registran como fármacos28, ya que, para estos dispositivos, el LNG no sólo concurre, sino que es el principal y único causante del efecto anticonceptivo18,35. El dispositivo sólo se encarga de controlar su liberación incidiendo en su eficacia y seguridad ciertamente más no ejerciendo por sí mismo alguna acción anticonceptiva30. Distinto a lo que ocurre con la T con cobre, por ejemplo, donde el marco y alas de cobre, es decir parte del material con el que está elaborado el dispositivo, es el que genera el efecto que finalmente lleva a la anticoncepción18,35.
Consideraciones FinalesEl DIU-LNG se considera una opción anticonceptiva reversible y muy eficaz. Se espera que los equivalentes genéricos de estos productos entren en el proceso de revisión y aprobación regulatoria en un futuro cercano. Por lo tanto, es fundamental que se disponga de métodos de evaluación de bioequivalencia in vitro/ex vivo e in vivo adecuados para garantizar la calidad y seguridad del producto.
Demostrar la bioequivalencia de los anticonceptivos de acción prolongada, incluyendo los DIU durante varios años de uso puede ser difícil y costoso. Sin embargo, aunque no se considera una terapia curativa o profilaxis, resulta muy relevante cautelar su eficacia y seguridad, dado el uso crónico y la relevancia de la prevención de embarazos no deseados.
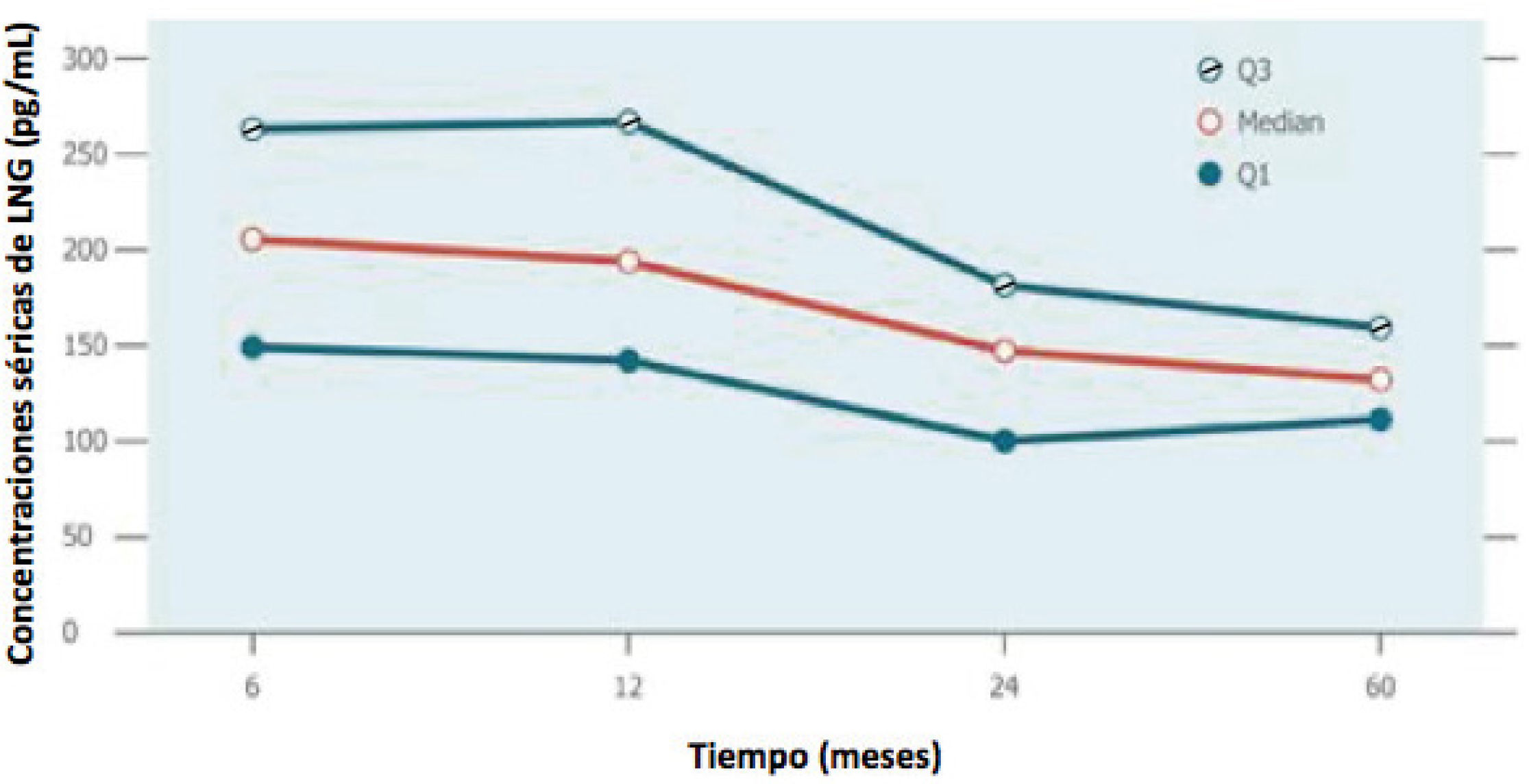
Un estudio de bioequivalencia in vivo de concentraciones séricas de al menos 12 meses, parece ser una alternativa factible de análisis comparativo dado el perfil de liberación establecido para el innovador (Figura 4) y puede acortar significativamente el tiempo de desarrollo del producto y también podría fomentar la competencia.

Perfiles de liberación de LNG por Mirena®
Q1: primer cuartil, Q3: tercer cuartil. Adaptado desde el Folleto de Información al profesional Mirena®, Bayer20.
Alternativa y complementariamente, algunos métodos in vitro podrían cumplir apropiadamente con los requerimientos de bioequivalencia, si cumplen estrictas condiciones de análisis. Sin embargo, las propiedades mecánicas únicas, el diseño del dispositivo y las características farmacológicas de los DIU-LNG innovadores presentan desafíos importantes para el desarrollo de genéricos. Por lo tanto, la ingeniería inversa y la caracterización precisa del DIU, el desarrollo de un método apropiado de liberación de fármacos in vitro, la determinación de estudios para establecer la bioequivalencia in vivo y el diseño apropiado del insertador deben considerarse en el desarrollo de un DIU-LNG genérico.
Las pruebas de liberación in vitro deben solicitarse previo al estudio in vivo, pues son una herramienta importante que no sólo garantiza la calidad del producto y el rendimiento constante, sino que también ayuda en el desarrollo del producto y los procesos de revisión regulatoria. Estas pruebas se han recomendado como parte de la demostración de bioequivalencia por la FDA8. Sin embargo, la falta de métodos actuales de liberación in vitro y la limitada información de la literatura para este tipo de dispositivos ha sido un obstáculo importante para el desarrollo de productos y la revisión regulatoria. Resulta por lo tanto muy relevante impulsar el desarrollo de métodos de liberación/disolución in vitro robustos y apropiados (tanto en tiempo real como acelerados) que puedan discriminar los DIU-LNG para predecir su rendimiento in vivo. Ciertamente que resulta obvio que para una formulación de DIU-LNG 52mg, Mirena® debe ser el referente de elección, por ser el primer DIU en liberar la restricción de patentes y por su respaldo científico.
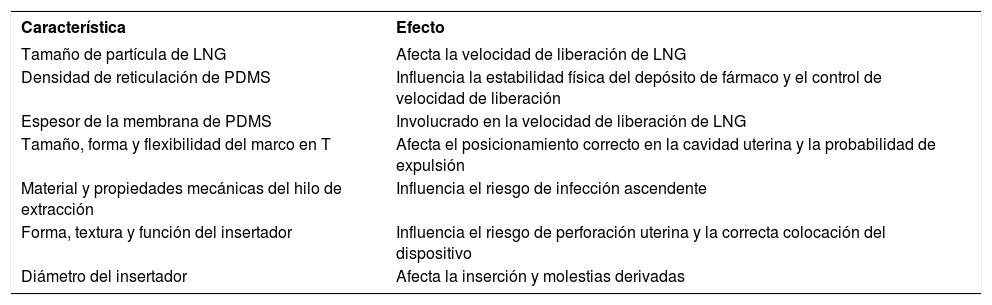
En el desarrollo de un DIU genérico, es fundamental caracterizar completamente el producto similar para garantizar que el producto tenga atributos de calidad clave comparables y, por lo tanto, un rendimiento in vivo similar. Las características fisicoquímicas principales que afectan la seguridad y eficacia del dispositivo y que deberían abordarse se presentan en la Tabla 4. Un solicitante de genérico debería presentar una justificación para demostrar que cualquier diferencia de diseño en comparación con el insertador del innovador no afecta la administración del producto por parte del proveedor de atención médica.
Características físico-químicas que afectan la seguridad y eficacia del DIU-LNG
| Característica | Efecto |
|---|---|
| Tamaño de partícula de LNG | Afecta la velocidad de liberación de LNG |
| Densidad de reticulación de PDMS | Influencia la estabilidad física del depósito de fármaco y el control de velocidad de liberación |
| Espesor de la membrana de PDMS | Involucrado en la velocidad de liberación de LNG |
| Tamaño, forma y flexibilidad del marco en T | Afecta el posicionamiento correcto en la cavidad uterina y la probabilidad de expulsión |
| Material y propiedades mecánicas del hilo de extracción | Influencia el riesgo de infección ascendente |
| Forma, textura y función del insertador | Influencia el riesgo de perforación uterina y la correcta colocación del dispositivo |
| Diámetro del insertador | Afecta la inserción y molestias derivadas |
La idea de los estudios comparativos in vitro es obtener una comprensión completa del efecto de las diferencias de fabricación sobre las propiedades físicas y mecánicas críticas de los DIU-LNG y su rendimiento in vitro e in vivo para el establecimiento de recomendaciones de bioequivalencia para los DIU-LNG genéricos y así proporcionar al público productos genéricos seguros y eficaces a un costo reducido y de manera oportuna.