


Activated Phospho-Inositide 3 (PI3) Kinases Delta syndrome (APDS) can underlie primary immune deficiency. The prevalence and phenotypic characterization of these patients are not well described in Egypt.
ObjectivesTo describe patients with APDS in hospitalized children with recurrent respiratory tract infections with suspected primary immune deficiency.
Methods79 patients were included in the study. E1021K and E525K mutations of PI3K δ chain gene were screened by Sanger sequencing technique.
Resultsone patient was heterozygous to E1021K mutation; a female child was diagnosed clinically as Combined Immune Deficiency with CD4 and B lymphopenia and markedly deficient IgG and increased IgM. The E525K mutation was not detected in our cohort.
ConclusionsScreening for APDS in patients with recurrent respiratory tract infections with undefined antibody deficiency or combined immune deficiency with or without bronchiectasis is required. These patients need great attention to benefit from the available treatment. Further studies on the Egyptian population are recommended to increase the knowledge about the prevalence and phenotypic characterization of this disease in Egypt.
Congenital or primary immune deficiency disorders (PIDs) are a heterogeneous group of diseases characterized by defects in the development and/or functions of immune system components, resulting not only in recurrent infections but also in immune dysregulation in the form of allergy, autoimmunity, and malignancy.1 According to the 2017 International Union of Immunological Societies (IUIS) phenotypic classification of primary immune deficiency, there are 320 gene defects underlying 330 phenotypic disorders.2 Over the last decade, the knowledge of PIDs and their molecular bases, as well as the ability to treat has been clearly growing. Therefore, early diagnosis of PIDs is essential to improve the overall survival of patients.3
Class 1A phospho-inositide 3 (PI3) kinases are heterodimers of a 110 KDa catalytic subunit whether (p110 α, p110 β or p110 δ) and a regulatory subunit p85 s. PI3Kα and β are expressed in all tissues and cells, while PI3Kδ is enriched mainly in leukocytes.4 These enzymes phosphorylate the 3 position of the inositol lipid of cell membrane generating phosphatidylinositol-3, 4, 5-triphosphates PI (3,4,5)P3. The generated PI (3,4,5) P3 recruits Akt to be activated by PDK1 and mTORC2.5,6
Class IA PI3K/AKT/mTOR signaling pathway has a crucial role in immune cell function mediation. p85 or p110δ knockout mice have functional defects in Neutrophils, Macrophages, Dendritic Cells, and Natural Killer cells even though these cells show normal development. Conversely, B cell development, B Cell Receptors (BCR) mediated proliferation, as well as antibody production are all impaired. In addition, T cell responses to cognate antigens are also impaired.7
Gain of function mutations in the p110δ gene were first described in 2013.6,8 These mutations in the catalytic subunit lead to increased activity of the PI3KCD, resulting in immune dysregulation and immunodeficiency causing a disease named Activated PI3K-δ Syndrome (APDS).8 Recently, up to 10 gain of function mutations in PI3KCD chain gene have been described.9 The most frequently reported one is the E1021K variant. It is an autosomal dominant missense Guanine (G) to Adenine (A) mutation at position c.3061. It encodes an amino-acid substitution "glutamic acid with lysine" at position 1021 (E1021K) in the C-lobe of the delta chain protein.8 Another one (E525K), is G to A mutation at position c.1573 with the same consequent amino acid substitution at position 525 in the helical domain, this substitution likely interferes with the inhibitory contacts between p110δ and p85α.10
Recently, loss of function mutations in the PIK3 regulatory chain (PIK3R1) gene have been described to cause increased PI3KCD activity and an immune deficiency disorder phenotypically resembling patients with the PIK3δ catalytic chain mutations. This disorder has been termed APDS2. Now, a PID caused by activating mutations in the PIK3δ catalytic chain gene is referred to as APDS1 and both diseases together are known as APDS.9
Activated PI3K-delta syndrome is characterized by recurrent respiratory tract infections, bronchiectasis, lymphoproliferation, poor antibody responses, expansion of senescent CD8 + T cells, B cell lymphopenia with relatively increased transitional B cell numbers, and reduced immunoglobulin IgG.6 APDS patients have been described to exhibit benign and malignant lymphoproliferative disease, often in association with EBV viremia.8 T cell lymphopenia was explained by the enhanced signaling of T Cell Receptors (TCR) leading to Activation Induced Cell Death (AICD). The defective B cell function was attributed to the suppression of class switching recombination while the increased transitional cell types is due to blocked B cell maturation or enhanced death of mature B.8
Recurrent respiratory tract infections in children ≥4 times per year are considered a warning sign of an underlying immunodeficiency disorder. APDS is represented mostly with recurrent pneumonia and bronchiectasis. The early diagnosis and correct treatment of these patients allows them to benefit from the commercially available inhibitor of PIK3δ.
Objectives and aim of this workTo evaluate the existence and to describe the phenotypic characterization of APD syndrome among children presented with recurrent respiratory tract infections at Zagazig University Hospital, to highlight the importance of screening for this disorder among Egyptian children with suspected PID.
Patients and methodsThis cross-sectional study was conducted at the pediatrics and clinical pathology departments of Zagazig University Hospital, Zagazig City, Al Sharqia Governorate, Egypt, from the first of July 2017 to January 2019. During the planning for this work and because the phenotypic characters of APDS patients had not been described before in Egypt, we preferred to widen the spectrum of the research to include children with recurrent respiratory tract infections with suspected PID. Subjects were selected from children admitted to the chest unit at the pediatrics department with severe or recurrent upper or lower respiratory tract infections with and without complications. Eligibility selection criteria included; age ≤ 16 from both genders, of the same Egyptian ethnicity, suspected to have a PID either by; 1) positive family history, 2) presence of any warning sign of immunodeficiency11 suspected from history taking and clinical examination, or 3) presence of any routine laboratory test results that indicate immunodeficiency such as lymphopenia. Written informed consents were obtained from parents to allow their child’s participation in the study.
Children with secondary immunodeficiency or whose parents refused to give informed consent were excluded from the study.
Data collectionAll selected patients were subjected to full history taking and thorough clinical examination. Results of imaging techniques including; chest X ray ± Computerized Tomography (CT) or Magnetic Resonance Imaging (MRI), routine laboratory investigations including; complete Blood Count (CBC), C Reactive Protein (CRP), serum immunoglobulin levels, lymphocyte subsets assessment by flowcytometry "for T cells (CD3,CD4 and CD8) and for B cells (CD19)" and specific laboratory investigations such as Nitro Blue Tetrazolium test, CD11 and CD18 assessment and CH50 assay test "if present" were collected from patients’ files.
Sample size calculationThe sample size of this study was calculated using this formula.12
where p is the prevalence of these mutations in a previous study = 0.008 and d is the absolute error or precision = 0.05.Ethical approvalThe Institutional Review Board (IRB) and ethical committee of Zagazig University Hospitals approved this study (Approval number: IRB#4704/26-6-2017). All subjects gave written informed consent before enrollment in this work. This study was conducted in accordance with the Declaration of Helsinki.
Genotyping of PI3KCD (p110 δ) for E1021K and E525K mutations by sanger sequencing1) DNA extraction; 2 ml whole blood on EDTA were collected, extraction was performed by Thermo Fisher Kits (Thermo Fisher Scientific Inc., Ontario, Canada) according to the manufacturer’s guidelines, 2) PCR amplification using the following primers; for E1021 K (forward 5’GAG ATG CTG GGA GCT CTC TACT3’- reverse5’TGT CGG TTC TTT CCC GTT AG3’).8
For E525 K (forward 5’CTA CCA GGC ATA TCT GGG GCC TTC3’- reverse 5’AGC TCC GCC CCC AGG TGC3’).13 PCR amplification reaction mixture contained 10 μl of the ready master mix (TOPsimple™PreMIX-nTaq, enzynomics, Daejeon, South Korea), 1 μl of 5 pmol from each primer, 2 μl of genomic DNA, and 6 μl nuclease free water, in a thermal cycler (Biometra TProfessional PCR, Göttingen, Germany) with cycling condition of initial denaturation at 95 °C for 5 min, 35 cycles of (denaturation at 95 °C for 30 s, annealing at 60 °C for 60 s and extension at 72 °C for 1 min), followed by a final extension at 72 °C for 5 min. On 1.5% agarose gel; amplicon bands that appeared at 380 and 590 bp for E1021 K and E525 K, respectively, were considered positive. 3) First purification of amplified PCR products with MEGAquick-spin™plus (INTRON Biotechnology, South Korea). 4) Cycle sequencing using Bigdye Terminator V3.1cycle sequencing kit (Thermo Fisher Scientific Inc., Ontario, Canada). 5) Second purification of the products using Centrisep spin column Purification Kit (Thermo Fisher Scientific Inc., Ontario, Canada). 6) Sequencing using the Applied Biosystems 3500 Genetic Analyzer (Thermo Fisher Scientific Inc., Ontario, Canada). All were performed according to the manufacturer’s guidelines. 7) The Nucleotide Blast online program (https://blast.ncbi.nlm.nih.gov) was used for results interpretation. Genotyping was performed at Zagazig university hospital research lab.
Further evaluation of positive casesPatients who were positive for any of the candidate mutations by DNA sequencing were subjected to further evaluation by: 1) Flowcytometry using the following markers from BD Bioscience (San Jose, USA); naive T cell (CD45RA), memory T cell (CD45RO), Transitional B cells (CD24 and CD38), memory B cell (CD27) and natural killer marker (CD56) using the BD FACScan™ flow cytometer (BD Bioscience, San Jose, USA), 2) Epstein Barr (EBV) and Cytomegalovirus (CMV) viremia by real time PCR using the LightCycler EBV Quant kit and LightCycler CMV Quant Kit with the LightCycler 2.0 Instrument (Roche Diagnostics, Mannheim, Germany).
Statistical analysisSPSS program version 21 (IBM Corp., Chicago, IL, USA) statistical software was used for data analysis. Shapiro–Wilk test was used to test the normality of quantitative results. Age and weight are expressed as median and range. Mann-Whitney test was used to compare medians. Frequencies are expressed as numbers and percentages, Chi-square test and Fisher’s exact test for results ≤5 were used to compare frequencies. P ≤ 0.05 was considered to indicate statistically significant differences.
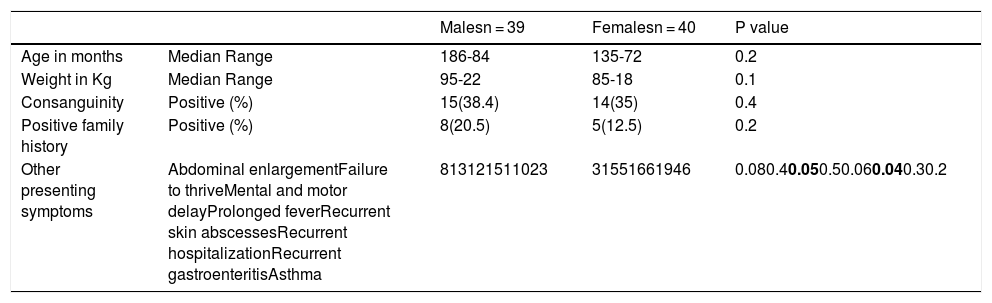
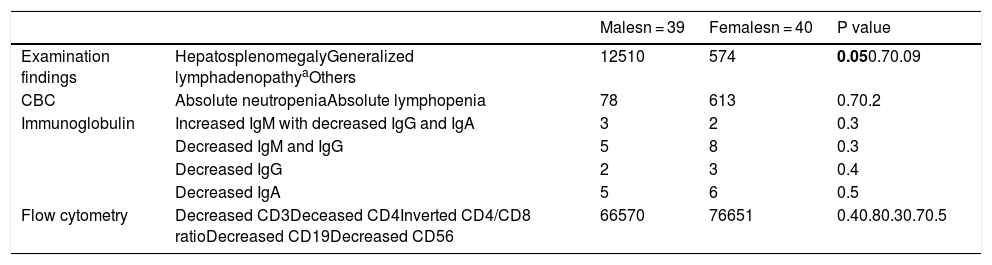
ResultsThis study included 82 children suspected to have a primary immune deficiency disorder. Three patients were excluded - one of them due to incomplete data and two due to failed sequencing. The remaining 79 children included 39 males and 40 females. Their median age was 15.5 months, range (5 months to 7 years) and median weight was 8 kg, range (5–22 kg). Demographic features, family history and presenting symptoms of the included patients are illustrated in Table 1. Signs and routine laboratory investigation results of patients are illustrated in Table 2. The main presenting symptom of patients was recurrent chest infections. Motor and mental delay, as well as hepatosplenomegaly, were more frequent in male children while the recurrent hospitalization was more frequent in female patients although the level of significance was weak p = 0.05 and 0.04, respectively.
Demographic features, family history, and presenting complaints of patients.
| Malesn = 39 | Femalesn = 40 | P value | ||
|---|---|---|---|---|
| Age in months | Median Range | 186-84 | 135-72 | 0.2 |
| Weight in Kg | Median Range | 95-22 | 85-18 | 0.1 |
| Consanguinity | Positive (%) | 15(38.4) | 14(35) | 0.4 |
| Positive family history | Positive (%) | 8(20.5) | 5(12.5) | 0.2 |
| Other presenting symptoms | Abdominal enlargementFailure to thriveMental and motor delayProlonged feverRecurrent skin abscessesRecurrent hospitalizationRecurrent gastroenteritisAsthma | 813121511023 | 31551661946 | 0.080.40.050.50.060.040.30.2 |
Significant P values are in bold.
Clinical signs and routine laboratory investigation results of patients.
| Malesn = 39 | Femalesn = 40 | P value | ||
|---|---|---|---|---|
| Examination findings | HepatosplenomegalyGeneralized lymphadenopathyaOthers | 12510 | 574 | 0.050.70.09 |
| CBC | Absolute neutropeniaAbsolute lymphopenia | 78 | 613 | 0.70.2 |
| Immunoglobulin | Increased IgM with decreased IgG and IgA | 3 | 2 | 0.3 |
| Decreased IgM and IgG | 5 | 8 | 0.3 | |
| Decreased IgG | 2 | 3 | 0.4 | |
| Decreased IgA | 5 | 6 | 0.5 | |
| Flow cytometry | Decreased CD3Deceased CD4Inverted CD4/CD8 ratioDecreased CD19Decreased CD56 | 66570 | 76651 | 0.40.80.30.70.5 |
Significant P values are in bold.
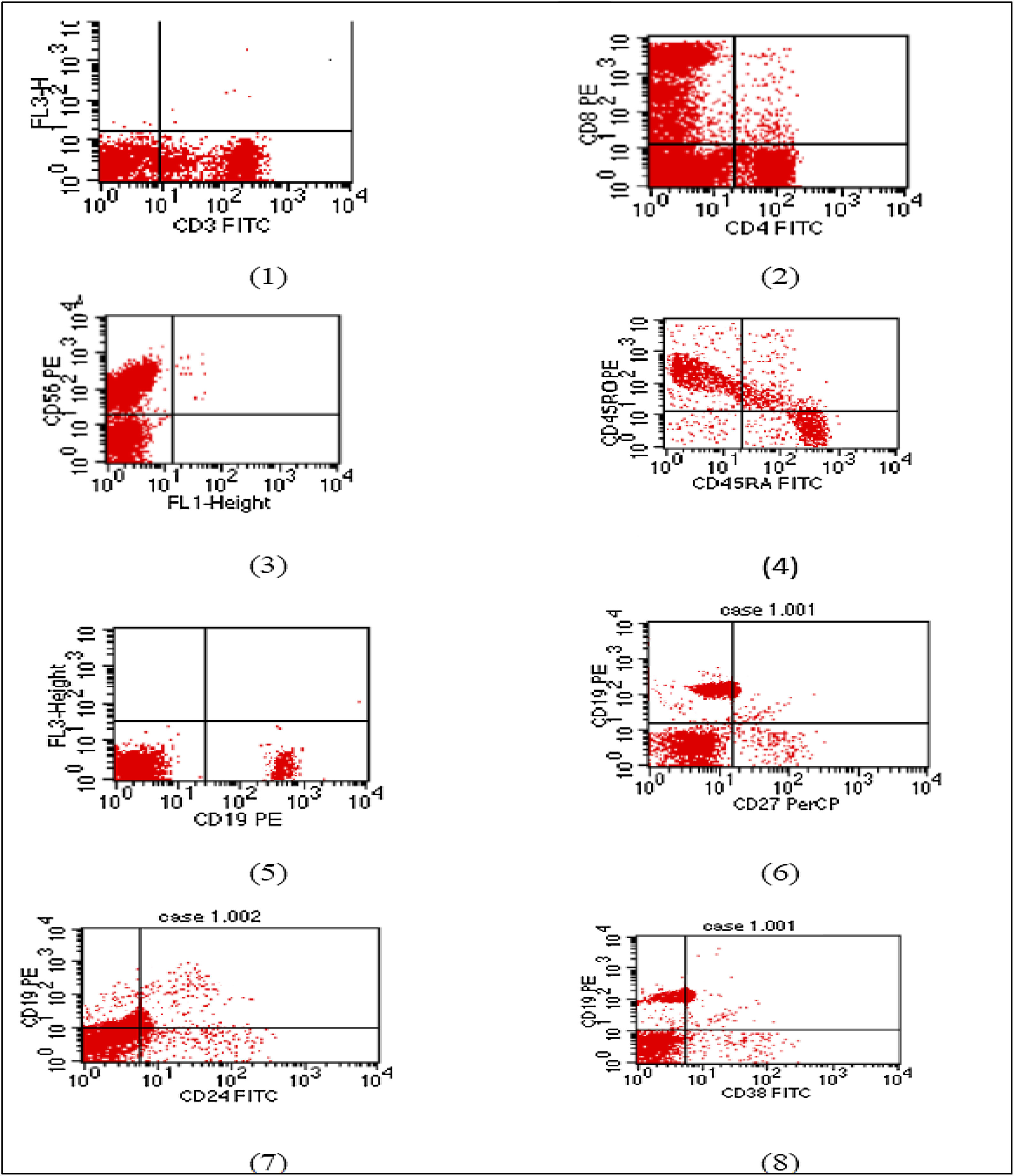
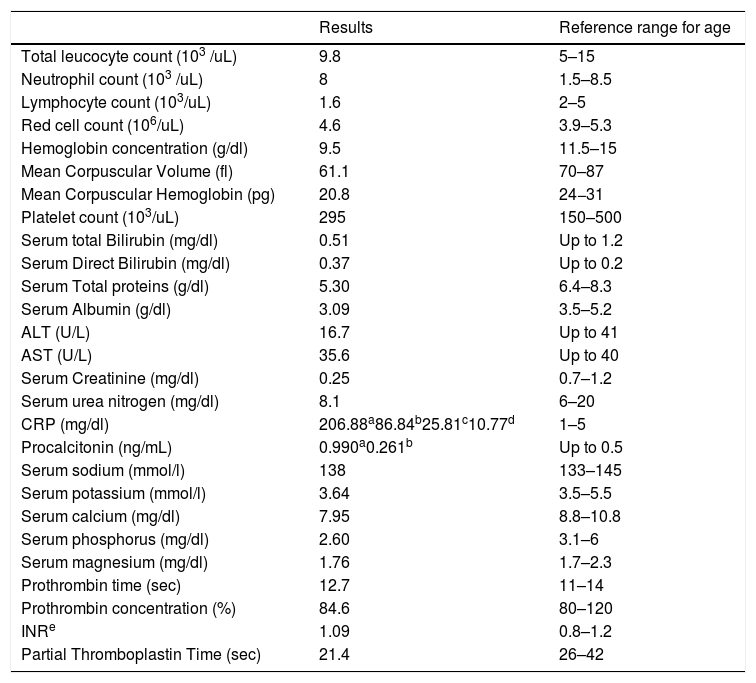
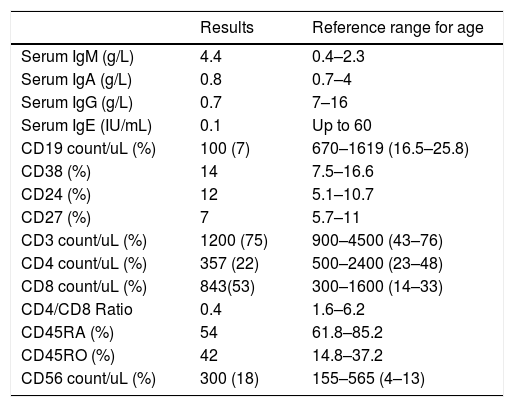
The E1021K positive patient referred to as (Patient 5) was a full term, 2.5 years old female, weight 9 kg, she was the second child in the family and the first child was healthy; there was positive consanguinity as the parents were cousins with a negative family history of PID. She had been admitted twice previously to hospital by recurrent chest infections at the age of 13 months and 19 months with the occurrence of frequent upper respiratory tract infections and sinusitis treated at home. This time she came to the hospital with difficulty breathing. In the examination, there were bilateral chest fine crepitations, cervical lymphadenopathy, no organomegaly, and no neurodevelopmental delay. The complete blood count of the patient showed normal total leukocyte count with marked lymphopenia, moderate microcytic hypochromic anemia, and normal platelet count. C reactive protein and Procalcitonine were highly increased in the first day of admission then decreased gradually with treatment. Flowcytometry for lymphocyte subsets showed the following; CD3 and CD8 were within the normal range, decreased CD4, inverted CD4/CD8 ratio with decreased CD45RA naive T cells and CD45RO were relatively increased. Markedly decreased CD19 with relatively increased transitional B cell (CD24 and CD38), normal CD56 were observed. Results of immunophenotyping are shown in Fig. 1. The immunoglobulin pattern showed markedly decreased IgG, normal IgA, and increased IgM. Laboratory investigation results of patient 5 as well as immunological investigation results are illustrated in Tables 3 and 4, respectively.
 CD3 within normal range (2) reduced CD4 with inverted CD4/CD8 ratio. (3) Natural killer cell within normal. (4) Reduced naïve T cell. (5) Markedly reduced B lymphocytes (6) memory B cells are within normal percentage. (7) and (8) Relatively increased transitional B cells.")
Flow cytometry charts of Patient 5.
(1) CD3 within normal range (2) reduced CD4 with inverted CD4/CD8 ratio. (3) Natural killer cell within normal. (4) Reduced naïve T cell. (5) Markedly reduced B lymphocytes (6) memory B cells are within normal percentage. (7) and (8) Relatively increased transitional B cells.
Laboratory investigation results of Patient 5 and reference ranges for age.
| Results | Reference range for age | |
|---|---|---|
| Total leucocyte count (103 /uL) | 9.8 | 5–15 |
| Neutrophil count (103 /uL) | 8 | 1.5–8.5 |
| Lymphocyte count (103/uL) | 1.6 | 2–5 |
| Red cell count (106/uL) | 4.6 | 3.9–5.3 |
| Hemoglobin concentration (g/dl) | 9.5 | 11.5–15 |
| Mean Corpuscular Volume (fl) | 61.1 | 70–87 |
| Mean Corpuscular Hemoglobin (pg) | 20.8 | 24−31 |
| Platelet count (103/uL) | 295 | 150–500 |
| Serum total Bilirubin (mg/dl) | 0.51 | Up to 1.2 |
| Serum Direct Bilirubin (mg/dl) | 0.37 | Up to 0.2 |
| Serum Total proteins (g/dl) | 5.30 | 6.4–8.3 |
| Serum Albumin (g/dl) | 3.09 | 3.5–5.2 |
| ALT (U/L) | 16.7 | Up to 41 |
| AST (U/L) | 35.6 | Up to 40 |
| Serum Creatinine (mg/dl) | 0.25 | 0.7–1.2 |
| Serum urea nitrogen (mg/dl) | 8.1 | 6–20 |
| CRP (mg/dl) | 206.88a86.84b25.81c10.77d | 1–5 |
| Procalcitonin (ng/mL) | 0.990a0.261b | Up to 0.5 |
| Serum sodium (mmol/l) | 138 | 133–145 |
| Serum potassium (mmol/l) | 3.64 | 3.5–5.5 |
| Serum calcium (mg/dl) | 7.95 | 8.8–10.8 |
| Serum phosphorus (mg/dl) | 2.60 | 3.1–6 |
| Serum magnesium (mg/dl) | 1.76 | 1.7–2.3 |
| Prothrombin time (sec) | 12.7 | 11–14 |
| Prothrombin concentration (%) | 84.6 | 80–120 |
| INRe | 1.09 | 0.8–1.2 |
| Partial Thromboplastin Time (sec) | 21.4 | 26–42 |
Immunological test results of Patient 5 and reference ranges for age.
| Results | Reference range for age | |
|---|---|---|
| Serum IgM (g/L) | 4.4 | 0.4–2.3 |
| Serum IgA (g/L) | 0.8 | 0.7–4 |
| Serum IgG (g/L) | 0.7 | 7–16 |
| Serum IgE (IU/mL) | 0.1 | Up to 60 |
| CD19 count/uL (%) | 100 (7) | 670–1619 (16.5–25.8) |
| CD38 (%) | 14 | 7.5–16.6 |
| CD24 (%) | 12 | 5.1–10.7 |
| CD27 (%) | 7 | 5.7–11 |
| CD3 count/uL (%) | 1200 (75) | 900–4500 (43–76) |
| CD4 count/uL (%) | 357 (22) | 500–2400 (23–48) |
| CD8 count/uL (%) | 843(53) | 300–1600 (14–33) |
| CD4/CD8 Ratio | 0.4 | 1.6–6.2 |
| CD45RA (%) | 54 | 61.8–85.2 |
| CD45RO (%) | 42 | 14.8–37.2 |
| CD56 count/uL (%) | 300 (18) | 155–565 (4–13) |
Epstein Barr Virus and Cytomegalovirus were negative. A chest X-ray showed bilateral bronchopneumonia, a CT scan revealed neither bronchiectasis nor mediastinal lymphadenopathy. Sputum culture and sensitivity were negative, most probably due to the previous administration of antibiotics before admission. The therapeutic regimen of this case consisted of antibiotic treatment with parenteral Ceftriaxone 75 mg/kg/day, vancomycin 15 mg/kg/dose/8 h, and fluconazole 9 mg/kg/day for 15 days and intravenous immunoglobulin therapy to resolve the concurrent infection with the planning for stem cell transplantation.
The final diagnoses of the involved patients were as follows: 21 (26.4%) patients had antibody deficiency, 11 (13.9%) of them had isolated IgA deficiency, five (6.3%) were diagnosed as Common Variable Immune Deficiency (CVID), three (3.7%) were diagnosed as hyper IgM syndrome, and two (2.5%) Bruton’s agammaglobulinemia. There were 11 (13.9%) patients with Combined Immune Deficiency (CID), two (2.5%) with Digeorge syndrome, two (2.5%) with Chediak-Higashi syndrome, one (1.3%) with Chronic Granulomatous disease, one (1.3%) with hyper IgE syndrome, one (1.3%) with adhesion molecules deficiencies, one (1.3%) with mitochondrial disorders, one (1.3%) with autoinflammatory disorders, one (1.3%) had C5 deficiency, and one (1.3%) Ataxia Telangiectasia syndrome. The remaining 36 (45.6%) patients were not confirmed to be PID patients, the causes of their recurrent respiratory infections were due to: bronchial asthma in nine (11.4%), gastro-esophageal reflux in six (7.6%), vitamin D deficiency in five (6.3%), inhaled foreign body in one patient (1.3%) and one case diagnosed as cystic fibrosis (1.3%). The remaining 14 (17.7%) patients were completely normal and the recurrent infections were attributed to environmental factors and poor hygiene.
DiscussionThis study was carried out in the chest unit of the pediatrics department at Zagazig university hospital. Seventy-nine children with recurrent respiratory tract infections were included; PID was proved in 43 patients (54.5%). The distribution of PID in these patients is compatible with registries in Egypt and other countries.14 Among the included 79 patients only one female patient (Patient 5) was heterozygous for E1021K mutation. This prevalence may be underestimating. Therefore, by excluding 36 patients with unproved PID and 11 patients with well-recognized syndromes of immune deficiency, the final prevalence of E1021 K mutation would be one in 32. The prevalence of APDS1 is not well established due to variations of the studied cohorts among different studies.9 Angulo et al. identified 17 APDS1 patients among 184 PID patients,8 this increased frequency was explained by the inclusion of multiple cases with hyper IgM syndrome, while Elgizouli et al. encountered five patients among 669 patients with undefined antibody deficiencies.13
The phenotypic characteristics of Patient 5 meet those of the previously described APDS patients in some aspects and differ in others. She was classified clinically as combined immune deficiency due to the deficient CD4 and B lymphocyte subsets with IgG deficiency with increased serum IgM. By returning to previous studies a wide heterogeneity in the immunological presentation of these cases was observed even within one family, with the predominance of recurrent respiratory tract infection in the majority of cases. Elgizouli et al. screened for four reported gain of function mutations in PI3K delta chain p110δ including (E1021K, E525K, N334K, and C416R) and one loss of function mutation in the PI3K regulatory chain p85α (c.1425 + 1). Only five patients were heterozygous for E1021K, three of them were siblings of one family and two were sporadic. The three siblings were presented as common variable immune deficiency (CVID) due to B lymphocytopenia and hypogammaglobulinemia, while the two sporadic cases were presented as combined immune deficiency due to B and T lymphopenia particularly CD4 and IgG deficiencies, with increased IgM in one of them whilst in the other it was normal.13 In a study by Crank et al.,15 they described three patients with APDS who presented with immunodeficiency and recurrent infection, with hyper reduced naive CD4 T cells and class-switched B lymphocytes, reduced IgG, IgA or IgE with increased serum IgM; they had been previously diagnosed as hyper IgM syndrome.
The E1021K mutation can be de novo and this can explain the negative family history of PID in Patient 5. Angluo et al. sequenced the healthy parents of an APDS patient and confirmed that they were homozygous for the normal allele.8 In addition, they found that the E1021K mutation is germline not somatic with any linkage disequilibrium to other genes; they also attributed the variability in the clinical presentation of patients to many factors including lifestyle, pathogens, treatment efficacy and possibly modifying genetic factors.
Activated PI3K-delta syndrome (APDS) patients are susceptible to complications such as airway damages. Bronchiectasis was described frequently in previous studies with variable incidence. Lucas et al.6 and Coulter et al.16 described bronchiectasis in 33% and 75% of their cohorts, respectively. These patients are prone to develop benign and malignant lymphoproliferation with and without association with EBV.6,15 Two of eight patients described by Kracker et al.17 developed B cell lymphoma. The young age of Patient 5 can explain the absence of lymphoproliferation as well as airway damages, since these complications may require time and recurrent episodes of infections particularly by herpes viruses to develop.16 Moreover, patients with APDS possess a high incidence to develop inflammatory and autoimmune diseases such as systemic lupus erythematosus.18 The severity of complications and high mortality rate signify the HSCT and the use of Rapamycin "the mTOR inhibitor".6
The E525K mutation was not detected in this study. It had, however, been identified in three patients out of 53 described with APDS1,16 yet according to Elgizouli et al., it has not been identified among the 669 patients with antibody deficiencies,13 indicating that E525K is much less common than E1021K.
The discovery of these mutations and their activating role has opened the opportunity for patients to gain benefit from selective PI3KCD inhibitors, which have been used as cancer chemotherapy19 and in inflammatory disorders.20–22 Idelalisib "a PI3KCD inhibitor approved for the treatment of chronic lymphocytic leukemia and non-Hodgkin lymphoma" reduces the catalytic activity of mutant PI3KCD efficiently to the level of the wild type.10 This opened the way for clinical trials on APDS1 patients. So, the screening for PI3KCD as well as PI3KR1 gene mutations in patients with atypical or undefined antibody deficiencies, bronchiectasis even without clear PID, severe herpes virus infection and lymphoma is recommended.16
Despite the increasing number of patients with APDS since the first description in 2013 and the rising importance as a cause of PID, the knowledge about this syndrome is not wide enough, as to the best of our knowledge no one in Egypt has studied this syndrome in PID patients, at least until the moment of writing this work. So, this study can be considered as a beginning to understand the prevalence and the clinical phenotype of these patients in our country. Because of this, during the plaining for this work we preferred to widen the spectrum of research to include children with suspected PID with recurrent respiratory tract infections, but in the light of our results and the results of previous literature worldwide we recommend to screen for APDS in children with recurrent infections who proved to have either undefined antibody deficiencies or CID with or without bronchiectasis, especially in infants and young children.
ConclusionScreening for PI3KCD as well as PI3KR1 gene mutations in recurrent respiratory tract infection patients with PID is required. APDS patients need great attention to gain benefit from the available treatment. In addition, further studies on a large cohort of Egyptian population are recommended to increase the knowledge about the prevalence and phenotypic characterization of APDS patients in Egypt.
LimitationsCell culture equipment and facilities for in vitro mitogen stimulation were not available. Parents would not allow us to try in vivo stimulation or take LN biopsy
Funding sourcesThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of interestAuthors have no conflict of interest to declare.
