


The prevalence of undiagnosed primary immunodeficiency diseases is remarkably high and contributes to increasing the rate of morbidity and mortality among this group of patients.
ObjectiveTo examine the 10 warning sign scoring system in patients suspected of primary immune deficiency and also estimate the diagnostic delay in patients with proven disease.
MethodsThis descriptive cross-sectional study was carried out during the years 2015–2016 in Ali Asghar (AS) Clinic and Hospital. Two hundred patients with suspected primary immune deficiency disease were eligible for inclusion in the study. Multivariable logistic regression analysis was used to determine the relation between findings.
ResultsIn this study, the majority of suspected cases of immunodeficiency were males (57%) with a mean age of 3.33±2.89 years. Twenty-one (10.5%) patients were diagnosed with immunodeficiency disease. The mean diagnostic delay among primary immune deficient patients was 2.05±1.7 years. There was a significant relationship between having parental consanguinity (OR=2.68, 95% CI: 1.07–6.70), allergies (OR=5.03, 95% CI: 1.13–22.31), vaccine adverse effects (OR=9.31, 95% CI: 1.24–69.96) and primary immune deficiency diagnosis. No association was observed between age (OR=0.98, 95% CI: 0.84–1.14), gender (OR=0.99, 95% CI: 0.39–2.47), immune deficiency scoring (OR=0.68, 95% CI: 0.31–1.45) and primary immune deficiency diagnosis.
ConclusionTen warning sign scoring system is of less value to consider a patient suspected of having primary immune deficiency. There is a meaningful delay in diagnosis of primary immune deficiencies especially in antibody deficiency defects which seeks further upgrading of knowledge in physicians.
Primary immune deficiency disorders (PID) are related to a group of disorders caused by a genetic defect in the immune system which makes the body susceptible to recurrent infections.1,2 Due to the wide spectrum of signs and symptoms with no clear relationship between clinical manifestations and the diagnosis, a multi-disciplinary approach is often required to diagnose and manage these disorders in a timely manner.3–5
Many countries have tried to create a database for PID.6–11 The results of these databases showed the geographical extent and racial differences in various types of PIDs. Iranian databases showed a prevalence of five to 10 cases per 100,000 people in Kerman province Study in 2006, while at the same time the record is more than 20 cases per 1,000,000 people in Esfahan, Tehran, Fars and Ghom Province.12 In a recent study carried out in Turkey, the prevalence of PID cases was 30.5 in 100,000 and predominantly diagnosed with antibody disorders.13 Other reports from Australia and New Zeeland have also showed a high prevalence rate of antibody defects that accounted for about 77% of cases.14 In addition, the worldwide prevalence rate of PID suggested that only 27,000–60,000 cases have been identified based on all national registries and the Jeffrey Modell Centers Network till 2012.15 It has been estimated that about 70–90% of individuals with a preexisting PID are still live without a definite diagnosis.16
Patients with untreated PID may suffer from life-threatening conditions such as infections, chronic organ injuries, and disabling disorders that could result in diminishing quality of life and life expectancy.16,17 Despite recent advancement in diagnostic modalities, early recognition of these disorders is still highly challenging and as a result a delay in diagnosis is frequently seen. Previous studies have also shown that an 8–24% delay in diagnosis is observed among PID patients.18,19 The delay in diagnosis is mainly due to insufficient knowledge of physicians about PIDs. Therefore, early diagnosis and proper treatment are the two key components to increasing survival and overall quality of life as well as limiting the substantially high economic costs to the healthcare system.20,21 Due to the importance of early diagnosis of PIDs, the present study was performed to evaluate patients suspected of PID and assess the amount of diagnostic delay and the utility of the Jeffery Model scoring system in the diagnosis of PIDs cases.
Material and methodsParticipantsThis cross-sectional study was performed in Ali-Asghar Pediatric Clinic and Hospital, a referral tertiary center in Tehran city. Two hundred patients who fulfilled at least one of our inclusion criteria were enrolled in this study over a period of two years. Patients were enrolled consecutively as they were referred to or needed to be consulted by the Pediatric Allergy and Immunology Specialist between October 2015 and October 2016. Patients were enrolled in the study according to one of the following inclusion criteria: 1. Six or more ear infections in one year, or two or more complications such as chronic tympanic perforation or mastoiditis. 2. Two or more serious sinus infection in one year. 3. Two or more pneumonias in one year. 4. Recurrent deep skin infections or internal organ abscess. 5. Severe skin or mucosal candidiasis (not including diaper rash or thrush). 6. Infection of unusual sites or with unusual organisms. 7. Two or more invasive infections such as sepsis, meningitis and osteomyelitis. 8. More than eight upper respiratory tract infections in one year. 9. Infections with poor response to antibiotic therapy. 10. Failure to thrive with no known origin. 11. Recurrent fever or fever persistent more than six weeks. 12. Family history of primary immune deficiencies or a history of sudden death in previous siblings. 13. Vaccine adverse effects. Patients with secondary immune deficiency (such as protein losing disorders, recent steroid therapy, with malignancies, etc.) and patients with structural abnormalities predisposing to infections were excluded from the study. Our exclusion criteria were defined as: 1. Malignancy or infiltrative disorders. 2. AIDS (acquired immune deficiency syndrome). 3. Protein loosing disorders. 4. Anatomic defects predisposing to recurrent infections. 5. Adverse or idiosyncratic drug effects. 6. Recent steroid or non-steroidal immunosuppressant use. 7. Known liver or kidney disease causing secondary immunodeficiency. 8. Anatomic or neurologic disorder leading to recurrent micro-aspirations and pneumonia. After a thorough examination, all participants were informed about their underlying condition. Treatment of diagnosed patients was conducted according to the national consensus regarding the management of PID patients.22 All personal identifiers have been removed from the data set. Written informed consent was obtained from all parents. The Ethics Committee of the university approved the consent form.
Study protocolMedical records of patients with a diagnosis of PID were reviewed by a physician to assess the consistency between clinical and laboratory evaluation with a given diagnosis, according to Practice Parameter for the Diagnosis and Management of Primary Immunodeficiency.23 Demographic information such as age were obtained from each patient. The past medical history of patients including data regarding recent hospital stay, parental consanguinity, family history of PID or sudden child death, allergies (including nasal and eye symptoms, allergic asthma, eczema, hives and anaphylaxis) and any history of vaccine adverse effects (e.g. pain, swelling, redness at the injection site, fever, irritability, drowsiness and BCGitis) or delayed umbilical cord separation were gathered. In order to reduce recall bias, we used the previous health records of each patient to attain precise information.
Each patient underwent specific laboratory tests to diagnose a possible underlying PID disease. A complete blood count was obtained from all patients. Serum immune globulins (Ig) concentrations including IgG, IgA, IgM, IgE and IgG subclasses and specific Ig on tetanus and diphtheria vaccination and isohemagglutinin test was performed using nephelometry. By means of flow cytometry, the cell markers for B-cells were established. For lymphopenic patients, a flow cytometry test was done to estimate the number of T-cell subtypes and T-cell subtype deficiencies. In addition, for phagocytic defects specific cell markers were obtained by means of flow cytometry and neutrophil reduction of dihydrorhodamine (DHR) and nitro blue tetrazolium chloride (NBT) was preformed via standard assays. Complement defects were studied by measuring CH50, C3 and C4 concentrations by nephelometry. The amount of diagnostic delay in patients diagnosed with PID was estimated by calculating the period between the time of first presenting signs and symptoms of PID and the time of diagnosis.
In order to determine the risk of PID, the Jeffery Model foundation's 10 warning signs of immune deficiency were used. The immune deficiency scoring criteria (IDR) were based on clinical signs and symptoms in patients with suspected underlying PID.24 These criteria provide 10 warning signs of PID and each patient is scored from 0 to 10. Patients with score two or higher are more likely to have an underlying PID. However, the 10 warning signs have shown some limitations due to failure in suggesting a risk of PID in high-risk patients.16,24 The 10 warning signs for immune deficiency are as follows: 1. Four or more ear infections in one year. 2. Two or more serious sinus infections in one year. 3. Two or more pneumonias in one year. 4. Recurrent, deep skin or organ abscesses. 5. Persistent thrush in mouth or fungal infection on skin. 6. Two or more deep-seated infections including septicemia. 7. Two or more months on antibiotics with little effect. 8. Need for intravenous antibiotics to clear infections. 9. Failure of an infant to gain weight or grow normally. 10. Family history of primary immunodeficiency.
Statistical analysisKolmogorov–Smirnov and Shapiro–Wilk tests were used to assess the data distribution. Quantitative variables were reported by mean±SD and percentage was used to describe qualitative variables. An independent sample T-test was used to compare quantitative variables, and Chi-square test was used to compare qualitative variables. Multiple logistic regression analysis was used to assess the association between PID and its related risk factors. All statistical analyses were performed using SPSS software version 23 (SPSS Inc.; Chicago, IL, USA). A P-value less than 0.05 was considered significant.
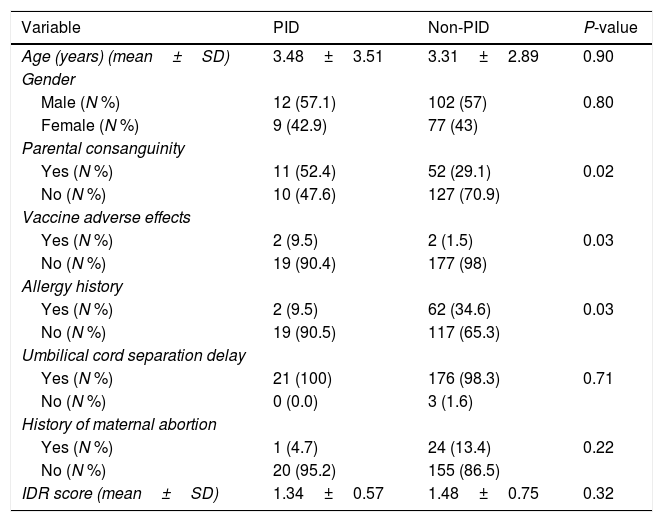
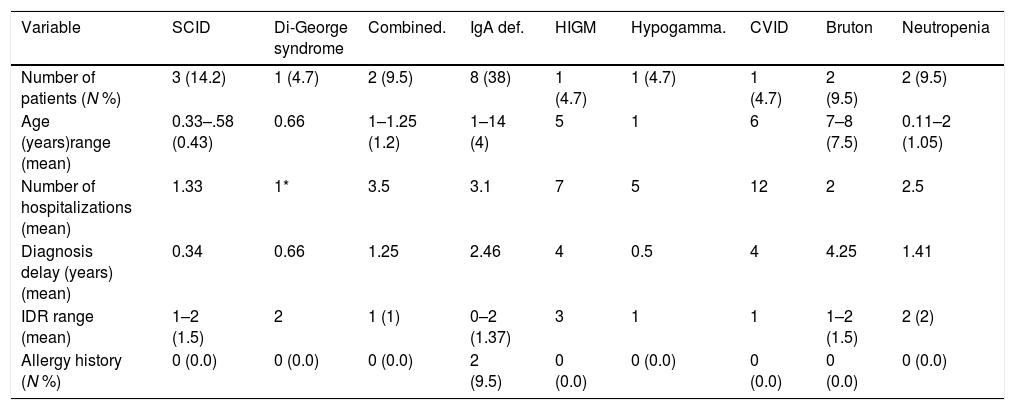
ResultsCharacteristics of patientsOf 200 patients enrolled in the study, 114 (57%) patients were males and 86 (43%) were females. Patients had an average age of 3.33±2.89 years. Among 200 patients, 195 (97.5%) were hospitalized and five (2.5%) cases were outpatients referred to the immunology clinic. Four patients had vaccine adverse effects of which two were diagnosed with PID (SCID). Three patients had a history of delayed umbilical cord separation but were not diagnosed to have PID. Sixty-three (31.5%) patients had parental consanguinity and 64 (32%) patients had a history of allergic disorders (Table 1). Fifty-six (28%) patients were diagnosed with pneumonia, 25 (12.5%) with sinusitis and 17 (8.5%) with otitis media. Of our 200 patients, 21 (10.5%) were diagnosed with PID; eight with IgA deficiency, three with severe combined immune deficiency (SCID), two with hypogammaglobulinemia, two with neutropenia, two with combined immune deficiency and one patient for each of common variable immune deficiency (CVID), hypogammaglobulinemia, Di-George and Hyper IgM syndrome (Table 2).
Descriptive statistics of patients.
| Variable | PID | Non-PID | P-value |
|---|---|---|---|
| Age (years) (mean±SD) | 3.48±3.51 | 3.31±2.89 | 0.90 |
| Gender | |||
| Male (N %) | 12 (57.1) | 102 (57) | 0.80 |
| Female (N %) | 9 (42.9) | 77 (43) | |
| Parental consanguinity | |||
| Yes (N %) | 11 (52.4) | 52 (29.1) | 0.02 |
| No (N %) | 10 (47.6) | 127 (70.9) | |
| Vaccine adverse effects | |||
| Yes (N %) | 2 (9.5) | 2 (1.5) | 0.03 |
| No (N %) | 19 (90.4) | 177 (98) | |
| Allergy history | |||
| Yes (N %) | 2 (9.5) | 62 (34.6) | 0.03 |
| No (N %) | 19 (90.5) | 117 (65.3) | |
| Umbilical cord separation delay | |||
| Yes (N %) | 21 (100) | 176 (98.3) | 0.71 |
| No (N %) | 0 (0.0) | 3 (1.6) | |
| History of maternal abortion | |||
| Yes (N %) | 1 (4.7) | 24 (13.4) | 0.22 |
| No (N %) | 20 (95.2) | 155 (86.5) | |
| IDR score (mean±SD) | 1.34±0.57 | 1.48±0.75 | 0.32 |
Hx: history; BCG: Bacillus Calmette-Guérin; PID: primary immune deficiency.
Descriptive statistics of immune deficiency patients.
| Variable | SCID | Di-George syndrome | Combined. | IgA def. | HIGM | Hypogamma. | CVID | Bruton | Neutropenia |
|---|---|---|---|---|---|---|---|---|---|
| Number of patients (N %) | 3 (14.2) | 1 (4.7) | 2 (9.5) | 8 (38) | 1 (4.7) | 1 (4.7) | 1 (4.7) | 2 (9.5) | 2 (9.5) |
| Age (years)range (mean) | 0.33–.58 (0.43) | 0.66 | 1–1.25 (1.2) | 1–14 (4) | 5 | 1 | 6 | 7–8 (7.5) | 0.11–2 (1.05) |
| Number of hospitalizations (mean) | 1.33 | 1* | 3.5 | 3.1 | 7 | 5 | 12 | 2 | 2.5 |
| Diagnosis delay (years) (mean) | 0.34 | 0.66 | 1.25 | 2.46 | 4 | 0.5 | 4 | 4.25 | 1.41 |
| IDR range (mean) | 1–2 (1.5) | 2 | 1 (1) | 0–2 (1.37) | 3 | 1 | 1 | 1–2 (1.5) | 2 (2) |
| Allergy history (N %) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (9.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
SCID: severe combined immune deficiency; Combined: combined immune deficiency; IgA Def: IgA deficiency; HIGM: Hyper IgM; Hypogamma: hypo-gammaglobulinemia.
Twenty-one patients were diagnosed with PID. Of these patients, 12 (57%) were male and nine (43%) were female (Table 2). None of the PID patients had a history of previous maternal abortion. No delayed umbilical cord separation was recorded (considering that none of our patients was diagnosed with leukocyte adhesive disorders). Two of our PID patients had vaccine adverse effects both diagnosed with SCID disorder, one with a history of BCGitis and the other SCID patient had an infected BCG vaccine site refractory to antibiotic therapy.
Eleven (52%) patients, including all SCID cases, Hyper IgM (HIGM), CVID, hypogammaglobulinemia, and combined immune deficient patients had parental consanguinity. The first most common complication among PID patients was pneumonia (n=10) followed by acute otitis media (n=5), prolonged fever without any proven origin, diarrhea and sinusitis. Patients with HIGM (n=7) and CVID (n=12) were more commonly hospitalized. The highest diagnostic delay was seen in patients with HIGM, hypogammaglobulinemia and CVID, all considered as humoral PID defects.
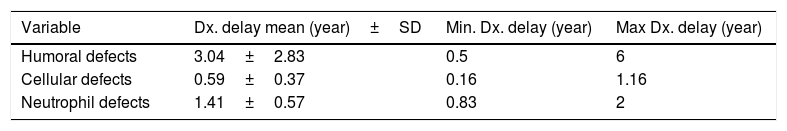
The mean IDR score was 1.59 for all patients. Two of our IgA patients had an IDR of 0. Six patients had an IDR of 1, three of whom were diagnosed with either SCID or combined immune deficiency. The highest IDR was among patients with HIGM syndrome and was found to have a diagnostic delay of four years (Table 2). Diagnostic delay was also analyzed based on the subclass of immune deficiency disorder (Table 3). Accordingly, most diagnostic delay was seen in humoral defects (mean=3.04 years) followed by neutropenic defects (mean=1.41). Cellular defects had the lowest diagnostic delay (mean=0.59).
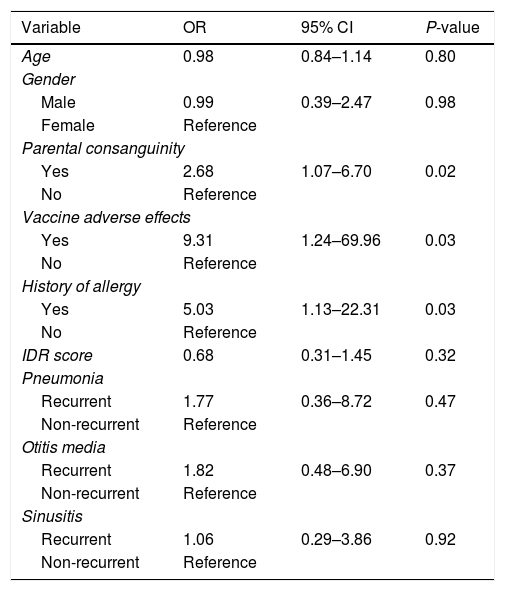
Association between PID and the study variablesLogistic regression analysis was used to determine the association between PID and the study variables (Table 4). There was a positive correlation between PID diagnosis and parental consanguinity (OR=2.68, 95% CI: 1.07–6.70), vaccine adverse effects (OR=9.31, 95% CI: 1.24–69.96) and positive allergy history (OR=5.03, 95% CI: 1.13–22.31). Comparison between the non-PID and PID group revealed that there was no positive correlation between age (OR=0.98, 95% CI: 0.84–1.14), gender (OR=0.99, 95% CI: 0.39–2.47) or IDR score (OR=0.68, 95% CI: 0.31–1.45) and established PID diagnosis. The association between PID diagnosis and variables including recurrent pneumonia, recurrent otitis media or recurrent sinusitis were as follows: recurrent pneumonia: OR=1.77, 95% CI: 0.36–8.72; recurrent otitis media: OR=1.82, 95% CI: 0.48–6.90; recurrent sinusitis: OR=1.06, 95% CI: 0.29–3.86. Although no positive correlation between these disorders and PID was observed, based on the odds ratio values, there was a significant association between having a recurrent respiratory infection and being diagnosed with PID.
Logistic regression for assessing association between primary immune deficiency and its risk factors.
| Variable | OR | 95% CI | P-value |
|---|---|---|---|
| Age | 0.98 | 0.84–1.14 | 0.80 |
| Gender | |||
| Male | 0.99 | 0.39–2.47 | 0.98 |
| Female | Reference | ||
| Parental consanguinity | |||
| Yes | 2.68 | 1.07–6.70 | 0.02 |
| No | Reference | ||
| Vaccine adverse effects | |||
| Yes | 9.31 | 1.24–69.96 | 0.03 |
| No | Reference | ||
| History of allergy | |||
| Yes | 5.03 | 1.13–22.31 | 0.03 |
| No | Reference | ||
| IDR score | 0.68 | 0.31–1.45 | 0.32 |
| Pneumonia | |||
| Recurrent | 1.77 | 0.36–8.72 | 0.47 |
| Non-recurrent | Reference | ||
| Otitis media | |||
| Recurrent | 1.82 | 0.48–6.90 | 0.37 |
| Non-recurrent | Reference | ||
| Sinusitis | |||
| Recurrent | 1.06 | 0.29–3.86 | 0.92 |
| Non-recurrent | Reference | ||
OR: odds ratio; CI: confidence interval.
Of 200 patients enrolled in this study, 21 (10.5%) were diagnosed with primary immune deficiency; eight with IgA deficiency, three with severe combined immune deficiency (SCID), two with hypogammaglobulinemia, two with neutropenia, two with combined immune deficiency and one patient for each of common variable immune deficiency (CVID), hypogammaglobulinemia, Di-George and Hyper IgM syndrome. In comparison between the non-PID and the PID group, there was no positive correlation between ages or gender and having a PID defect (OR=0.98, 95% CI: 0.84–1.14 for age and OR=0.99, 95% CI: 0.39–2.47 for gender).
The most common defect diagnosed was IgA deficiency (66%), followed by SCID disorder (14%). A study on 930 patients of the Iranian PID Registry in 20069 showed that antibody deficiencies were the most common form of PID in Iranian patients (38.4%), followed by congenital defects of phagocyte number and/or function (28.3%), other well-defined immunodeficiency syndromes (17.7%), and combined T- and B-cell immunodeficiency (11.0%), complement deficiencies (2.4%), and diseases of immune dysregulation (2.3%). Among antibody deficiency disorders, CVID was the most common disorder followed by IgA deficiency (18). The difference between the results of this study and our study may be due to different statistical populations, and also may be a result of increased diagnosis of PIDs in recent years in Iran. Thus, a new study on the prevalence of PIDs in the Iranian population is needed.
In another study in southern Sweden conducted on 259 patients suspected with PID, 15 patients were diagnosed with PID. The most common defect was CVID, found in eight patients, followed by hypogammaglobulinemia. The inclusion criteria were similar to our study except that they did not enroll patients with failure to thrive of an unknown cause. Another difference was that they had nine patients who could be classified as transient hypogammaglobulinemia of infancy or unclassified hypogammaglobulinemia but were not included as PID patients in the final analysis.19 In the present study, the most common PID diagnosed was IgA deficiency which requires long-term follow-up in case these patients develop another antibody deficiency disorder such as CVID. Another reason why the CVID prevalence was low in our study is due to two ranges of age peaks for symptoms of CVID. One at 5–6 years of age and another at 20–30 years.25 The second period was not involved in our study, thus justifying the lower prevalence of CVID.
In a study conducted on 100 patients suspected of having PID in Egypt in 2008, analysis on B and T-cell defects found 35 patients with PID; the most common defect found was CVID followed by IgA deficiency and T-cell defects of no definite diagnosis.26 Our study results on the prevalence of the type of PID diagnosed resembled another study in Egypt in the period of July 2011–July 2013 on patients suspected of PID, where the most common PID identified was IgA deficiency, followed by SCID disorder.27 The most common PID diagnosed in our study was Ab body deficiency disorder, compatible with most centers and the global study of PID performed by the Jeffery Modell Foundation with 51.6% Ab deficiency cases reported.28
Our study shows a positive relationship between parental consanguinity and the risk of having a PID disorder (OR=2.68, 95% CI: 1.07–6.70). About half of our PID patients had parental consanguinity (Tables 1 and 4). All patients with SCID, combined disorders, CVID, Bruton's disease, hypogammaglobulinemia, and one patient with IgA deficiency had consanguineous parents. The results were comparable with another study on 515 Iranian patients in 2006 in which the largest number of consanguineous marriages were reported in the parents of patients with cellular and combined deficiencies.9 In a retrospective study (1984–2010) on 93 CVID patients in Iran, 72.4% of patients had consanguineous parents.29 In another study in Egypt the history of consanguineous marriage was 62.5%.30 In contrast, in another study in Mexico, the consanguinity rate was only 3.85%, which might be due to specific genetic characteristics of patients with PID in the Mexican population.31
All patients were scored 0–10 based on the 10 warning signs of immune deficiency described by the Jeffery Model foundation and some other non-profit foundations.24 Of our PID patients, eight patients had an IDR of 1, and two patients had an IDR of 0. Patients with an IDR of 1 were diagnosed with SCID, combined disorders, CVID, hypogammaglobulinemia and IgA deficiency. Two of our IgA-deficient patients had an IDR of 0. There was no positive relationship between IDR score and being diagnosed with PID (OR=0.68, 95% CI: 0.31–1.45). Therefore, relying on the 10 warning signs to predict the susceptibility to PIDs is not beneficial and might miss some cases of PID. These results were compatible with the results of the Egyptian research where 16% of the PID patients had a score of 1 or 0.27 Over the past few years, our understanding regarding the complexity of the immune system has dramatically evolved and several studies have shown that the current 10 warning signs are not sufficient to accurately detect the various presentations of PID – particularly in cases with less classical presentations or having a longer diagnostic delay. One of the reasons for a longer diagnostic delay in PID patients may be attributed to the growing emphasis on the 10 warning signs alone. It is important to note that training of clinicians from other specialties than immunology or infectious disease in immunodeficiency disorders is of great importance due to the diverse phenotypes of PID patients’ manifestations. An in-depth understanding of clinical presentations of PID that is related to their specialty is more warranted that referring to a simple list of warning signs. It is worth mentioning that factors including type of infection, affected organ, allergies and vaccine-related adverse effects should also be added to the list of PID warning signs. In addition, more studies from other parts of the world are strongly recommended as these epidemiological data could help in determining other clinical presentations of PID that have not been emphasized previously.
The most common complication among PID patients was pneumonia (n=10) followed by acute otitis media (n=5); and the third most common complication was prolonged fever without any proven origin (n=4). Diarrhea and sinusitis both held the fourth place (n=3). In a 2008 study in India, the most common infection in 27 PID diagnosed patients was pneumonia, followed by diarrhea and deep-seated abscess.32 In the Indian study there were four cases of chronic granulomatous disease, which explains the difference. Microbial agents that are commonly seen in these disorders include pneumonia, otitis media and sinusitis, thus explaining why these infections are of the most common infections seen in PID. But one should bear in mind other types of PIDs, uncommon organisms such as mycobacterium, Aspergillosis, Staphylococcus aureus, Pneumocystis jirovecii and others that may cause severe infections, causing infections at unordinary sites.33
We also evaluated the association between recurrent respiratory tract infections with PID diagnosis. The values for recurrent pneumonia (OR=1.77, 95% CI: 0.36–8.72), recurrent otitis media (OR=1.82, 95% CI: 0.48–6.90) and recurrent sinusitis (OR=1.06, 95% CI: 0.29–3.86) show that there is no significant correlation between PID diagnosis and these infections, but the results of the odds ratio do show an important association between them. As a result, one can argue that these infections are important factors influencing PID diagnosis, particularly in combination with other risk factors and conditions relevant with PID. We had no patients with delayed umbilical cord separation. Accordingly, none of our PID patients had a diagnosis of leukocyte adhesive disorder (LAD) of any type. There was a positive correlation between allergy and PID diagnosis (OR=5.03, 95% CI: 1.13–22.31). Two of our PID patients had a positive history of allergies, and both were among our IgA-deficiency patients. Studies showed that IgA-deficiency patients are more prone to allergic disorders.34 The most common allergies seen are asthma followed by allergic rhinitis and conjunctivitis.35
The mean diagnostic delay in our PID patients was 2.05±1.7 years and ranged from 0.16 to six years. Antibody deficiency defects had the longest diagnostic delay (mean=3.04 years) and cellular defects had the shortest diagnosis delay (mean=0.59 years) as described in Tables 2 and 3. This difference in delayed diagnosis between PID subgroups is expected because of different presentations of these disorders. Humoral disorders have milder signs and symptoms with a delayed onset of presentation. Their presentations are more similar to those diseases commonly seen in non-PID patients, such as sinusitis and otitis media but sometimes with a more severe course than usual. But in phagocytic and cellular defects the onset of presentations is usually in early life and are more severe in nature.
Delayed diagnosis of PID causes a great deal of morbidity and mortality. Severe or recurrent infections can cause mortality or permanent organ damage. Malignancies and autoimmune diseases are another complication of PIDs for which, if diagnosed earlier, adequate treatment might prevent them or delay their onset. Delayed diagnosis may be due to little awareness of non-immunologist physicians about PIDs, assuming recurrent infections as a normal pattern and not linking presentations other than infection to PIDs as an underlying cause.36–38 A study in Iran showed a mean delayed diagnosis of Ab deficiency defects of 2.11±1.87 years between 1997 and 2007.39 In our study the mean delayed diagnosis for these disorders was 3.04±2.38 years. The difference might be due to different sample sizes between the two studies. In 1994, a study on Ab deficiency disorders showed more than 10 years delay between the onset of clear symptoms and referral to an immunologist, which indicates significant improvement in PID diagnosis in recent years.40 In another study in the United Kingdom 1989–2002, the overall median delay for Ab deficiencies was two years (mean, 4.4) which showed an improvement in practice since the years before.41 Due to recent studies improvement in diagnosis is still a matter of concern and more attempts are needed to lower the rate of delayed diagnosis. This goal can be achieved by means of improving physicians’ awareness of PID disorders and their burden on patients and healthcare services.
A review on our patientsWe had three patients diagnosed with SCID. One was a 4.5-month-old boy with acute otitis media, oral aphthous and BCGitis. He had consanguineous parents and a history of three uncle deaths in the first year of their lives with unknown reason. Another patient was a seven-month-old boy with infection of BCG vaccination site resistant to antimicrobial therapy. Bacillus Calmette-Guérin vaccination is routinely administered to infants in Iran within the first month of life, usually at birth. BCGitis is a complication we might see in non-PID patients but along with other infections one should doubt if there is an underlying cause.33 The third patient with SCID was a four-month-old girl diagnosed with cytomegalovirus (CMV) pneumonia. CMV is an uncommon cause of pneumonia in otherwise healthy patients.
We had two patients with combined PID. Both of whom had low surface cell markers for T-helper cells and B-cells. One was a 14-month-old boy who had consanguineous parents. He had a twin sibling who died after suffering severe infections. Our patient was hospitalized with FTT, diarrhea and sepsis with Staphylococcus epidermidis. The other patient was a 15-month-old girl with CMV pneumonia and autoimmune hemolytic anemia. Her parents have a consanguineous marriage and he had a sibling who died of unknown reason at a very young age.
Our patients with neutropenia were two patients: one an 11-month-old girl with persistent diarrhea and persistent oral thrush, and the other a two-year-old boy with recurrent oral ulcers and hospitalization due to septicemia. Both are receiving granulocyte-colony stimulating factor (GCSF) since the diagnosis which had improved their situation.
We had two cases of hypogammaglobulinemia, both of whom had parental consanguinity. One patient was a seven-year-old boy who had a leg infection when he was 1.5 years old (parents did not know the exact site of infection in the leg), recurrent oral aphthous since he was two, fevers of unknown origin since he was four and lung abscess in his 6th year of life. He was diagnosed with PID with a delay of four years. He was started with periodic IVIg injection. The other patient was an eight-year-old boy also with recurrent oral aphthous, and with recurrent otitis media, and with pneumonia at the time enrolled in this study. He was also started on IVIg injection. We had one case of CVID which was a boy aged six who had had recurrent fever of unknown origin since he was two years old and recurrent diarrhea since he was six months old. He had parents with a consanguineous marriage.
We had one case of CVID which was a boy aged six and had recurrent fever of unknown origin since he was two years old and recurrent diarrhea since he was two months old. He had parents with a consanguineous marriage.
Our type 5 hyper IgM patient was a five-year-old girl with recurrent sinusitis, recurrent complicated otitis media and recurrent conjunctivitis who suffered the infections since infancy and has had many hospitalizations and ventilation tube insertion in both ears. She had parental consanguinity. She was diagnosed with PID with a four-year delay. Thus, it denotes a great deal of cost for the patient and the health care system.
We had an eight-month-old patient who was hospitalized since his birth with patent ductus arteriosus (PDA), hypoplastic thymus, facial abnormalities, lymphopenia with low cellular surface markers for all T-cell subtypes. He has suffered recurrent pneumonias and has had a blood culture positive for Candida albicans, Acinetobacter baumannii and Pseudomonas aeruginosa. He was finally diagnosed with Di-George syndrome.
Our hypogammaglobulinemia patient was a one-year-old boy with a history of meningitis, pneumonia and recurrent diarrhea since he was six months old. He had low levels of IgM, IgA and a period of low IgG levels. If this is a case of transient hypogammaglobulinemia or not is a matter which should become clear in close follow up of his health condition and laboratory results.
We had eight patients whose primarily diagnosis was of selective IgA deficiency. They had an age range between one and 14 years old. The main complications among them were recurrent sinusitis, recurrent otitis media, recurrent fevers, and pneumonia. Two of the patients did not fulfill the 10 warning signs of PID. As some IgA deficient patients may progress to become common variable immune deficiency (CVID) in future, thus close follow-up of these patients is needed.
LimitationsThis study was performed in Ali Asghar Hospital, which is a referral tertiary care center of pediatric diseases. As a result, the results on PID prevalence in this study cannot be generalized to Iran's population. Moreover, more specific tests for the diagnosis of PID including memory B cells, specific antibodies for pneumococcus, BTK, CD40L, IFN-IL12 axis and molecular study were lacking.
ConclusionWith improvement in physicians’ knowledge and laboratory tests, more cases of PID are being diagnosed worldwide. A more comprehensive education program is needed to improve the knowledge about PIDs to decrease the delay in diagnosis and thus, decrease the morbidity and mortality of patients and the health care system financial costs, respectively. The cornerstone of PID diagnosis is the presence of recurrent infectious disease, and thus family physicians as well as infectious diseases specialists are the first to encounter patients with a possible underlying immunodeficiency. Therefore, new strategies for increasing the knowledge of various specialties are warranted for better understanding of PID manifestations by these physicians. Moreover, to improve the timely diagnosis of PID, it is not only necessary to improve education on the subject, but also neonatal screening is required, implementing specialized laboratory techniques and networking with other centers as well. In addition, factors including type of infection, affected organ, allergies and vaccine-related adverse effects should also be added to the list of PID warning sign. Future larger studies are needed to propose a new diagnostic paradigm for early recognition of PID and reduce the diagnostic delay.
Conflict of interestNone declared.
The authors would like to thank Ali Asghar Clinical Research Development Center (AACRDC), Assistance.
