


Bacille Calmette-Guerin (BCG) vaccination has a great impact on the prevention of severe complications of tuberculosis. However, in patients with primary immunodeficiencies (PID), it can lead to severe complications such as severe combined immunodeficiency, chronic granulomatous disease, and Mendelian susceptibility to mycobacterial disease. This study highlights the demographics, clinical complications and laboratory parameters among PID patients associated with BCG vaccination side effects.
MethodsOne hundred and thirty-seven PID patients with BCGosis were evaluated in this study, based on the complications following BCG vaccination.
ResultsThe mean age of the patients with BCG complications at the time of the first visit was five years. The within-group comparison of patients showed a highly significant incidence of pneumonia and hepatomegaly in severe combined immunodeficiency patients. Furthermore, the immunologic data showed an increase in the overall rates of lymphocytes such as CD3+, CD4+ and CD8 + T cells in Mendelian susceptibility to mycobacterial disease patients. The level of immunoglobulins has also increased in chronic granulomatous disease patients.
ConclusionThe high rate of undiagnosed PIDs predisposes individuals to a high risk of severe side effects as a result of BCG vaccination, as well as infants that are less than one month of age. Therefore, there is a need for early screening and diagnosis of PIDs before exposing unknown PID status patients to BCG vaccination. The benefits of screening and early diagnosis of PID cannot be overemphasized, especially in patients with a previous family history of immunodeficiency.
Bacillus Calmette–Guérin (BCG) vaccine is a live attenuated strain of Mycobacterium Bovis, which is routinely administered to neonates to prevent early-life serious infections with Mycobacterium Tuberculosis, especially in countries with a high prevalence of tuberculosis (TB). The precise efficacy and protection of the BCG vaccine are unclear. However, a comprehensive review of the literature has shown that the BCG vaccine prevents the occurrence of meningitis and miliary tuberculosis in children by 64–78%.1 The Canadian vaccination guide suggests BCG vaccination for areas of the world in which TB incidence is more than 1%, however, the use of the BCG vaccine is not completely safe.2 Currently, BCG vaccination is ongoing as part of the routine national immunization program in more than 170 countries (http://www.bcgatlas.org).
Naturally, BCG vaccination in immunocompetent neonates leads to the manifestation of 5–15 mm red indurated area in the injection site. However, localized lymphadenopathy may manifest in the absence of localized inflammation evidence, such as redness and vesicles.3,4 Overall, localized complications manifest between 0.1 to 19 per 1,000 vaccinations, and serious complications such as disseminated tuberculosis are almost 1:1,000,000 vaccinated persons, usually associated with immune system defects.3,5 BCG vaccine administration in patients with primary immunodeficiency disorders (PIDs) can cause a potentially lethal infection, especially in those who suffer from severe combined immunodeficiency (SCID), Mendelian susceptibility to mycobacterial diseases (MSMD) and chronic granulomatous disease (CGD).6–8 Other types of PIDs with a genetically heterogeneous background affecting different compartments of the immune system, including syndromic and non-syndromic CID, can also present BCG complications.9–11
Given the potential impact of complications following the BCG vaccine in patients with PIDs, we evaluated BCGosis in our national registry of PID patients, and we compared demographic data, clinical manifestations and immunological findings of the three main PID disorders associated with these complications.
Material and methodsPatients with side effects of BCG complication from 1983 to 2019 12 and under clinical follow up based on the national consensus management for PID 13 at the Children's Medical Center (Pediatrics Center of Excellence affiliated to Tehran University of Medical Sciences, Tehran, Iran) were included in this retrospective study. The clinical diagnosis of PID was established according to the newest European Society for Immunodeficiency (ESID) criteria.14 Secondary defects of the immune system, including those caused by human immunodeficiency virus (HIV) were ruled out. Laboratory evaluations were performed in the study group as indicated, including complete blood counts and differential count, serum protein profile and immunoglobulin (Ig) levels, serum IgG subclass levels, isohemagglutinin titers, specific antibody responses, disease-specific autoantibody measurements, flow cytometric evaluation of lymphocyte subsets, nitro blue tetrazolium dye (NBT) test, lymphocyte transformation and T-cell function tests. Microbiological, pathological and imaging evaluations were performed in case of medical indication for clinical diagnosis.12
A questionnaire was designed that contained all patients’ information, such as demographic data (including age at the time of study, age at onset, age at diagnosis, delay of diagnosis, family history of PID), clinical manifestations, laboratory data, vaccination, and its complication and therapeutic profile. A positive history of BCG vaccination, as well as two or more signs of BCGosis including fever, loss of weight, lymphadenopathy, abscess near or outside of the lymph nodes, pneumonia, hepatosplenomegaly, and other sign of mycobacterial infection, were considered for determining BCGosis. In this regard, BCGosis complications were classified as localized or disseminated. Based on a normal range of T and B cells in Tables 1 and 2, we considered T- or B- for values less than the normal range and T + or B + for values within the normal range.
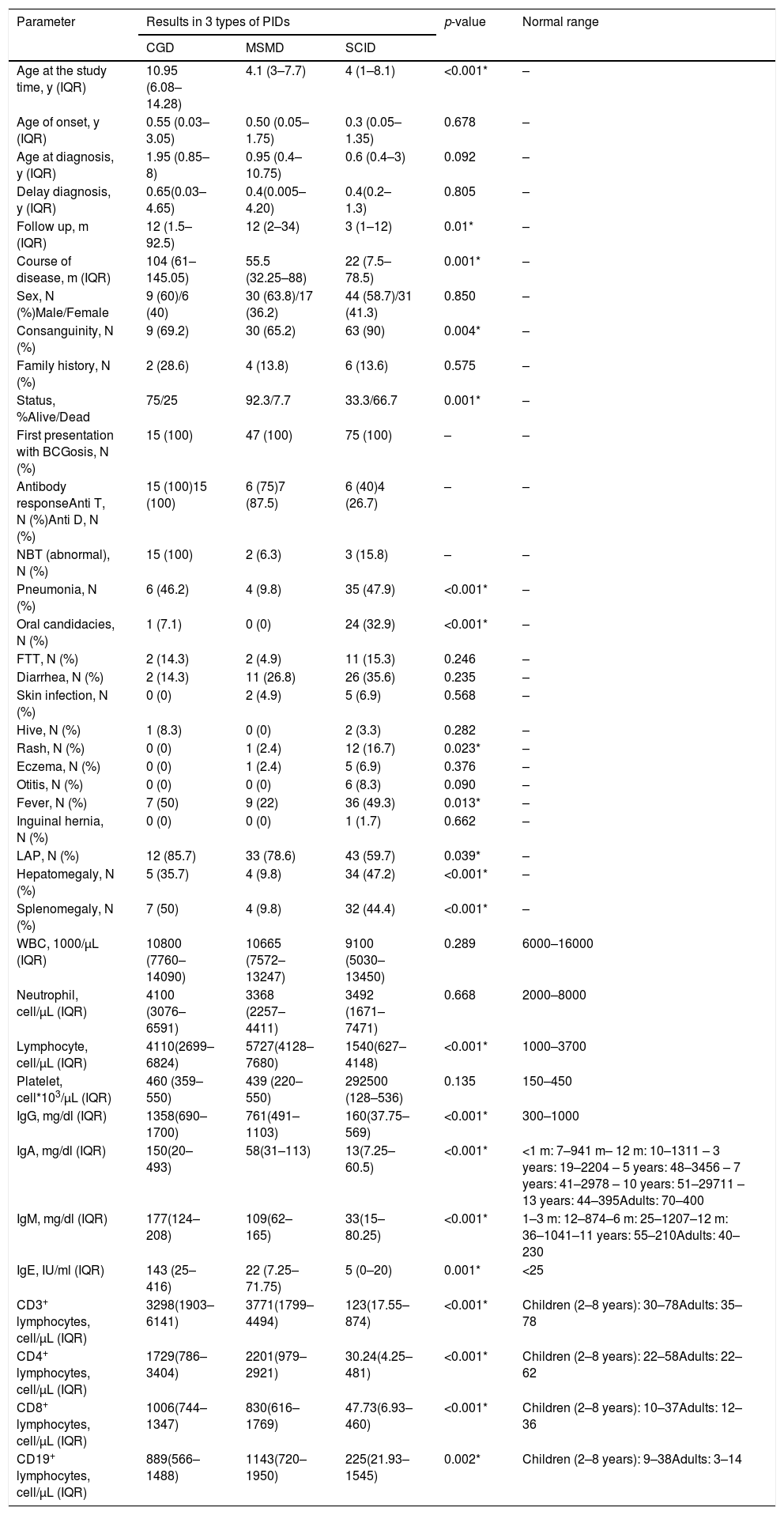
Comparison of demographic, clinical and laboratory data between three groups of patients with CGD, MSMD, and SCID.
| Parameter | Results in 3 types of PIDs | p-value | Normal range | ||
|---|---|---|---|---|---|
| CGD | MSMD | SCID | |||
| Age at the study time, y (IQR) | 10.95 (6.08–14.28) | 4.1 (3–7.7) | 4 (1–8.1) | <0.001* | – |
| Age of onset, y (IQR) | 0.55 (0.03–3.05) | 0.50 (0.05–1.75) | 0.3 (0.05–1.35) | 0.678 | – |
| Age at diagnosis, y (IQR) | 1.95 (0.85–8) | 0.95 (0.4–10.75) | 0.6 (0.4–3) | 0.092 | – |
| Delay diagnosis, y (IQR) | 0.65(0.03–4.65) | 0.4(0.005–4.20) | 0.4(0.2–1.3) | 0.805 | – |
| Follow up, m (IQR) | 12 (1.5–92.5) | 12 (2–34) | 3 (1–12) | 0.01* | – |
| Course of disease, m (IQR) | 104 (61–145.05) | 55.5 (32.25–88) | 22 (7.5–78.5) | 0.001* | – |
| Sex, N (%)Male/Female | 9 (60)/6 (40) | 30 (63.8)/17 (36.2) | 44 (58.7)/31 (41.3) | 0.850 | – |
| Consanguinity, N (%) | 9 (69.2) | 30 (65.2) | 63 (90) | 0.004* | – |
| Family history, N (%) | 2 (28.6) | 4 (13.8) | 6 (13.6) | 0.575 | – |
| Status, %Alive/Dead | 75/25 | 92.3/7.7 | 33.3/66.7 | 0.001* | – |
| First presentation with BCGosis, N (%) | 15 (100) | 47 (100) | 75 (100) | – | – |
| Antibody responseAnti T, N (%)Anti D, N (%) | 15 (100)15 (100) | 6 (75)7 (87.5) | 6 (40)4 (26.7) | – | – |
| NBT (abnormal), N (%) | 15 (100) | 2 (6.3) | 3 (15.8) | – | – |
| Pneumonia, N (%) | 6 (46.2) | 4 (9.8) | 35 (47.9) | <0.001* | – |
| Oral candidacies, N (%) | 1 (7.1) | 0 (0) | 24 (32.9) | <0.001* | – |
| FTT, N (%) | 2 (14.3) | 2 (4.9) | 11 (15.3) | 0.246 | – |
| Diarrhea, N (%) | 2 (14.3) | 11 (26.8) | 26 (35.6) | 0.235 | – |
| Skin infection, N (%) | 0 (0) | 2 (4.9) | 5 (6.9) | 0.568 | – |
| Hive, N (%) | 1 (8.3) | 0 (0) | 2 (3.3) | 0.282 | – |
| Rash, N (%) | 0 (0) | 1 (2.4) | 12 (16.7) | 0.023* | – |
| Eczema, N (%) | 0 (0) | 1 (2.4) | 5 (6.9) | 0.376 | – |
| Otitis, N (%) | 0 (0) | 0 (0) | 6 (8.3) | 0.090 | – |
| Fever, N (%) | 7 (50) | 9 (22) | 36 (49.3) | 0.013* | – |
| Inguinal hernia, N (%) | 0 (0) | 0 (0) | 1 (1.7) | 0.662 | – |
| LAP, N (%) | 12 (85.7) | 33 (78.6) | 43 (59.7) | 0.039* | – |
| Hepatomegaly, N (%) | 5 (35.7) | 4 (9.8) | 34 (47.2) | <0.001* | – |
| Splenomegaly, N (%) | 7 (50) | 4 (9.8) | 32 (44.4) | <0.001* | – |
| WBC, 1000/µL (IQR) | 10800 (7760–14090) | 10665 (7572–13247) | 9100 (5030–13450) | 0.289 | 6000–16000 |
| Neutrophil, cell/µL (IQR) | 4100 (3076–6591) | 3368 (2257–4411) | 3492 (1671–7471) | 0.668 | 2000–8000 |
| Lymphocyte, cell/µL (IQR) | 4110(2699– 6824) | 5727(4128–7680) | 1540(627–4148) | <0.001* | 1000–3700 |
| Platelet, cell*103/µL (IQR) | 460 (359–550) | 439 (220–550) | 292500 (128–536) | 0.135 | 150–450 |
| IgG, mg/dl (IQR) | 1358(690–1700) | 761(491–1103) | 160(37.75–569) | <0.001* | 300–1000 |
| IgA, mg/dl (IQR) | 150(20–493) | 58(31–113) | 13(7.25–60.5) | <0.001* | <1 m: 7–941 m– 12 m: 10–1311 – 3 years: 19–2204 – 5 years: 48–3456 – 7 years: 41–2978 – 10 years: 51–29711 – 13 years: 44–395Adults: 70–400 |
| IgM, mg/dl (IQR) | 177(124– 208) | 109(62–165) | 33(15–80.25) | <0.001* | 1–3 m: 12–874–6 m: 25–1207–12 m: 36–1041–11 years: 55–210Adults: 40–230 |
| IgE, IU/ml (IQR) | 143 (25–416) | 22 (7.25–71.75) | 5 (0–20) | 0.001* | <25 |
| CD3+ lymphocytes, cell/µL (IQR) | 3298(1903–6141) | 3771(1799–4494) | 123(17.55–874) | <0.001* | Children (2–8 years): 30–78Adults: 35–78 |
| CD4+ lymphocytes, cell/µL (IQR) | 1729(786–3404) | 2201(979–2921) | 30.24(4.25–481) | <0.001* | Children (2–8 years): 22–58Adults: 22–62 |
| CD8+ lymphocytes, cell/µL (IQR) | 1006(744–1347) | 830(616–1769) | 47.73(6.93–460) | <0.001* | Children (2–8 years): 10–37Adults: 12–36 |
| CD19+ lymphocytes, cell/µL (IQR) | 889(566–1488) | 1143(720–1950) | 225(21.93–1545) | 0.002* | Children (2–8 years): 9–38Adults: 3–14 |
Abbreviations: Ig: Immunoglobulins; CD: Cluster of Differentiation; y: years. D: Diphtheria; T: tetanus; NBT: Nitro blue tetrazolium; WBC: White Blood Cells; FTT: Failure To Thrive; LAP: Lymphadenopathy.
Note. For quantities data, the median is shown [with IQR, 25th and 75th percentiles]. N, Count.
*p-value <0.05 has been regarded as significant.
Comparison of demographic, clinical and laboratory data between four phenotypes of patients with SCID.
| Parameter | T-B- | T-B+ | p-value | Normal range | ||
|---|---|---|---|---|---|---|
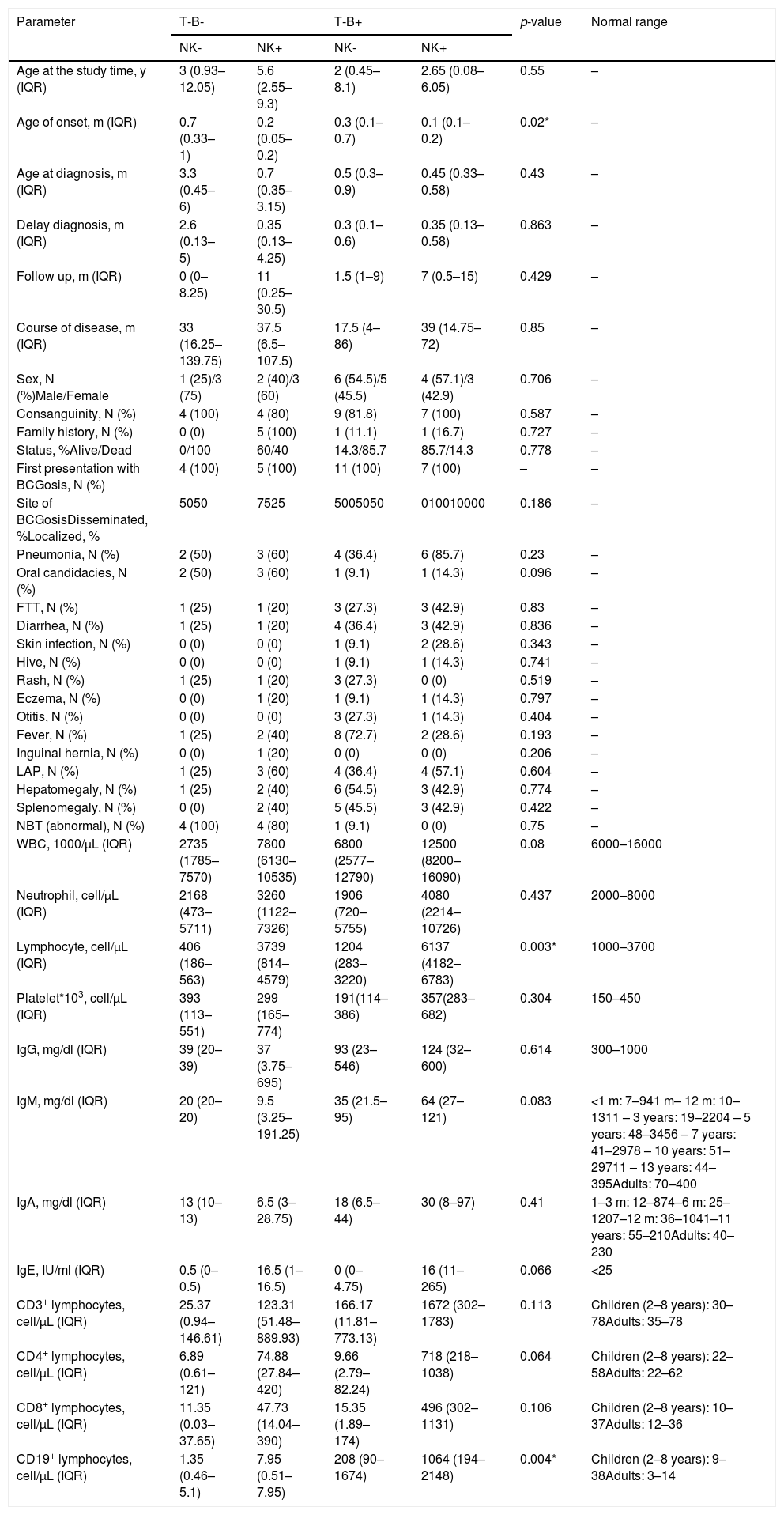
| NK- | NK+ | NK- | NK+ | |||
| Age at the study time, y (IQR) | 3 (0.93–12.05) | 5.6 (2.55–9.3) | 2 (0.45–8.1) | 2.65 (0.08–6.05) | 0.55 | – |
| Age of onset, m (IQR) | 0.7 (0.33–1) | 0.2 (0.05–0.2) | 0.3 (0.1–0.7) | 0.1 (0.1–0.2) | 0.02* | – |
| Age at diagnosis, m (IQR) | 3.3 (0.45–6) | 0.7 (0.35–3.15) | 0.5 (0.3–0.9) | 0.45 (0.33–0.58) | 0.43 | – |
| Delay diagnosis, m (IQR) | 2.6 (0.13–5) | 0.35 (0.13–4.25) | 0.3 (0.1–0.6) | 0.35 (0.13–0.58) | 0.863 | – |
| Follow up, m (IQR) | 0 (0–8.25) | 11 (0.25–30.5) | 1.5 (1–9) | 7 (0.5–15) | 0.429 | – |
| Course of disease, m (IQR) | 33 (16.25–139.75) | 37.5 (6.5–107.5) | 17.5 (4–86) | 39 (14.75–72) | 0.85 | – |
| Sex, N (%)Male/Female | 1 (25)/3 (75) | 2 (40)/3 (60) | 6 (54.5)/5 (45.5) | 4 (57.1)/3 (42.9) | 0.706 | – |
| Consanguinity, N (%) | 4 (100) | 4 (80) | 9 (81.8) | 7 (100) | 0.587 | – |
| Family history, N (%) | 0 (0) | 5 (100) | 1 (11.1) | 1 (16.7) | 0.727 | – |
| Status, %Alive/Dead | 0/100 | 60/40 | 14.3/85.7 | 85.7/14.3 | 0.778 | – |
| First presentation with BCGosis, N (%) | 4 (100) | 5 (100) | 11 (100) | 7 (100) | – | – |
| Site of BCGosisDisseminated, %Localized, % | 5050 | 7525 | 5005050 | 010010000 | 0.186 | – |
| Pneumonia, N (%) | 2 (50) | 3 (60) | 4 (36.4) | 6 (85.7) | 0.23 | – |
| Oral candidacies, N (%) | 2 (50) | 3 (60) | 1 (9.1) | 1 (14.3) | 0.096 | – |
| FTT, N (%) | 1 (25) | 1 (20) | 3 (27.3) | 3 (42.9) | 0.83 | – |
| Diarrhea, N (%) | 1 (25) | 1 (20) | 4 (36.4) | 3 (42.9) | 0.836 | – |
| Skin infection, N (%) | 0 (0) | 0 (0) | 1 (9.1) | 2 (28.6) | 0.343 | – |
| Hive, N (%) | 0 (0) | 0 (0) | 1 (9.1) | 1 (14.3) | 0.741 | – |
| Rash, N (%) | 1 (25) | 1 (20) | 3 (27.3) | 0 (0) | 0.519 | – |
| Eczema, N (%) | 0 (0) | 1 (20) | 1 (9.1) | 1 (14.3) | 0.797 | – |
| Otitis, N (%) | 0 (0) | 0 (0) | 3 (27.3) | 1 (14.3) | 0.404 | – |
| Fever, N (%) | 1 (25) | 2 (40) | 8 (72.7) | 2 (28.6) | 0.193 | – |
| Inguinal hernia, N (%) | 0 (0) | 1 (20) | 0 (0) | 0 (0) | 0.206 | – |
| LAP, N (%) | 1 (25) | 3 (60) | 4 (36.4) | 4 (57.1) | 0.604 | – |
| Hepatomegaly, N (%) | 1 (25) | 2 (40) | 6 (54.5) | 3 (42.9) | 0.774 | – |
| Splenomegaly, N (%) | 0 (0) | 2 (40) | 5 (45.5) | 3 (42.9) | 0.422 | – |
| NBT (abnormal), N (%) | 4 (100) | 4 (80) | 1 (9.1) | 0 (0) | 0.75 | – |
| WBC, 1000/µL (IQR) | 2735 (1785–7570) | 7800 (6130–10535) | 6800 (2577–12790) | 12500 (8200–16090) | 0.08 | 6000–16000 |
| Neutrophil, cell/µL (IQR) | 2168 (473–5711) | 3260 (1122–7326) | 1906 (720–5755) | 4080 (2214–10726) | 0.437 | 2000–8000 |
| Lymphocyte, cell/µL (IQR) | 406 (186–563) | 3739 (814–4579) | 1204 (283–3220) | 6137 (4182–6783) | 0.003* | 1000–3700 |
| Platelet*103, cell/µL (IQR) | 393 (113–551) | 299 (165–774) | 191(114–386) | 357(283–682) | 0.304 | 150–450 |
| IgG, mg/dl (IQR) | 39 (20–39) | 37 (3.75–695) | 93 (23–546) | 124 (32–600) | 0.614 | 300–1000 |
| IgM, mg/dl (IQR) | 20 (20–20) | 9.5 (3.25–191.25) | 35 (21.5–95) | 64 (27–121) | 0.083 | <1 m: 7–941 m– 12 m: 10–1311 – 3 years: 19–2204 – 5 years: 48–3456 – 7 years: 41–2978 – 10 years: 51–29711 – 13 years: 44–395Adults: 70–400 |
| IgA, mg/dl (IQR) | 13 (10–13) | 6.5 (3–28.75) | 18 (6.5–44) | 30 (8–97) | 0.41 | 1–3 m: 12–874–6 m: 25–1207–12 m: 36–1041–11 years: 55–210Adults: 40–230 |
| IgE, IU/ml (IQR) | 0.5 (0–0.5) | 16.5 (1–16.5) | 0 (0–4.75) | 16 (11–265) | 0.066 | <25 |
| CD3+ lymphocytes, cell/µL (IQR) | 25.37 (0.94–146.61) | 123.31 (51.48–889.93) | 166.17 (11.81–773.13) | 1672 (302–1783) | 0.113 | Children (2–8 years): 30–78Adults: 35–78 |
| CD4+ lymphocytes, cell/µL (IQR) | 6.89 (0.61–121) | 74.88 (27.84–420) | 9.66 (2.79–82.24) | 718 (218–1038) | 0.064 | Children (2–8 years): 22–58Adults: 22–62 |
| CD8+ lymphocytes, cell/µL (IQR) | 11.35 (0.03–37.65) | 47.73 (14.04–390) | 15.35 (1.89–174) | 496 (302–1131) | 0.106 | Children (2–8 years): 10–37Adults: 12–36 |
| CD19+ lymphocytes, cell/µL (IQR) | 1.35 (0.46–5.1) | 7.95 (0.51–7.95) | 208 (90–1674) | 1064 (194–2148) | 0.004* | Children (2–8 years): 9–38Adults: 3–14 |
Abbreviations: Ig: Immunoglobulins; CD: Cluster of Differentiation; y: years; D: Diphtheria; T: tetanus; NBT: Nitro blue tetrazolium; WBC: White Blood Cells; FTT: Failure To Thrive; LAP: Lymphadenopathy.
Note. For quantities data, the median is shown [with IQR, 25th and 75th percentiles]. N, Count.
*p-value <0.05 has been regarded as significant.
Informed consent was obtained from all patients. This study was approved by the Medical Ethics Committee of Tehran University of Medical Sciences (IR.TUMS.REC.1395.2761).
Statistical analysisData analysis was performed using SPSS software (SPSS Statistics 17.0.0; SPSS, Chicago, IL, USA). Our data were analyzed by non-parametric tests such as Kruskal–Wallis and Mann–Whitney test. Kaplan-Meier curve was used to estimate the survival rate in SCID patients. Pearson Chi-Square and Fisher’s exact test were used for comparing our groups. A p-value of less than 0.05 was considered statistically significant. Data were shown as an absolute number, percentages, median and interquartile range (IQR).
ResultsDemographic profileIn this study, 137 patients (39.4% females) with BCG vaccine complications were evaluated. Among these patients, 75 patients were diagnosed with SCID, followed by 47 patients with MSMD and 15 patients with CGD. BCGosis were observed in only 29.7% out of the total 460 patients who were diagnosed with SCID, MSMD, and CGD in our registry. We did not identify any other patients with CID, syndromic CID or immune dysregulation with BCG complication in this cohort.
Note that this study comprises patients that are mostly Indo-Europeans, and that 79.1% of the recruited 137 cases were from the consanguine parents, and 15.3% had a family history of immunodeficiency disorders. Furthermore, all the patients received vaccination at birth and the median age for the first referral to the clinic was five years (2.1-8.35 years old). The duration of follow up (p = 0.01) and the course of the disease were significantly shorter in SCID patients (p = 0.001) (Table 1). The age of onset was significantly lower in T-B + NK + SCID patients with a median of 0.1 (0.1-0.2) months (p = 0.02) Table 2).
Clinical presentationLymphadenopathy was recognized in all the patients following the routine BCG vaccination. The median age for the first lymphadenopathy manifestation was four months (at the birth to 1.5 years old). Furthermore, the involvement of spleen and liver was observed in 33.9% of patients, who had disseminated side effects following the vaccination.
The incidence of oral candidiasis (32.9%, p<0.001), dermatologic manifestations (16.7%, p = 0.023), and hepatomegaly in SCID patients (47.2%, p < 0.001) was significantly higher than other patients.
SCID and CGD patients were more frequently diagnosed with pneumonia and fever, compared to MSMD patients (p < 0.001 and p = 0.013, respectively). Note that patients diagnosed with SCID and CGD were more likely to develop splenomegaly (p < 0.001) and generalized lymphadenopathy (submandibular, supraclavicular, or intuitive, p < 0.001), whereas, in MSMD patients, lymphadenopathy was more localized (Table 1).
Immunologic laboratory profileSCID patients were categorized based on their phenotype into B- and B + group. In our cohort, 66.6% of SCID patients had B + phenotype and the rest of them demonstrated B- phenotype. Among these patients, the majority of them were T-B + NK- (40.7%), and 25.9% were T-B + NK+, 18.5% were T-B-NK + and 17.8% were T-B-NK- (Table 2). The severe BCG vaccine complications were seen in SCID patients with T-B + NK- phenotype.
The data obtained from flow cytometry showed that the lymphocyte counts in SCID patients have decreased, whereas these same cells have increased in CGD and MSMD patients (p < 0.001). In addition, the percentage of CD3+, CD4+, CD8+ lymphocyte has significantly increased in MSMD and CGD patients, while showing a significant reduction/absence in SCID patients (p < 0.001). Moreover, there was a significant difference in the percentage of CD19+ across the three groups of patients (increased in MSMD and decreased in SCID, p = 0.002). Evaluation of serum immunoglobulins showed increased levels of IgG, IgA, IgM, IgE in CGD patients, but decreased levels in SCID patients (p < 0.001) (Table 1).
Additionally, the total lymphocytes count and CD19+ lymphocytes were significantly lower in T-B-NK- SCID patients (p = 0.003 and p = 0.004, respectively) (Table 2). Other demographics, clinical and laboratory data were not significantly different among the four phenotypes of SCID patients (p > 0.05).
There was no defect regarding the anti-tetanus and anti-diphtheria immune response in CGD patients. However, impaired antibody production against tetanus and diphtheria toxoids was detected in a high proportion of SCID patients (60% and 73.3%, respectively). Data obtained from the NBT test showed that there was a defect in all the CGD patients, whereas it was normal in the MSMD and SCID patients, as expected. All laboratory information and their comparison among the three types of PIDs are shown in Table 1.
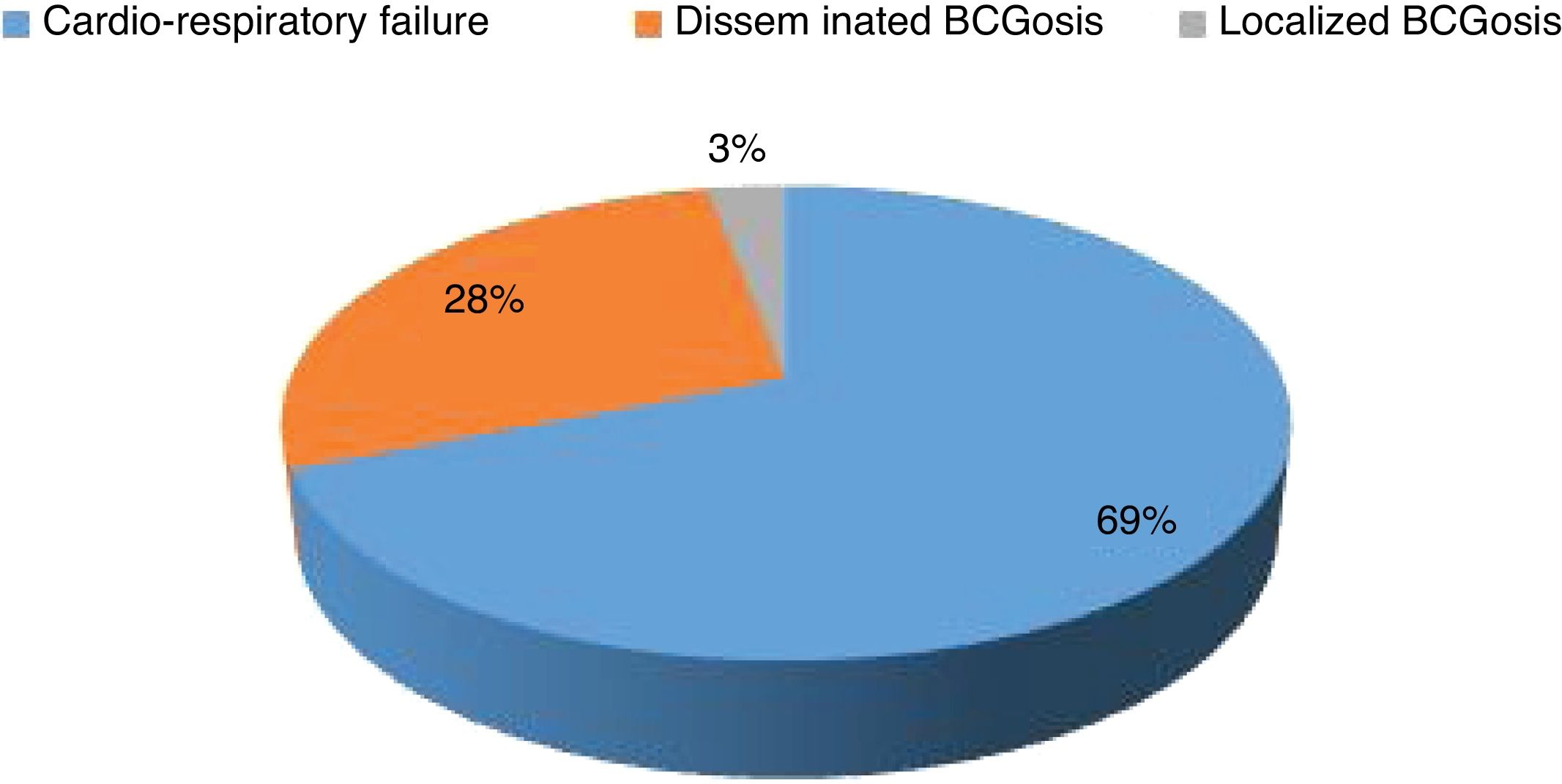
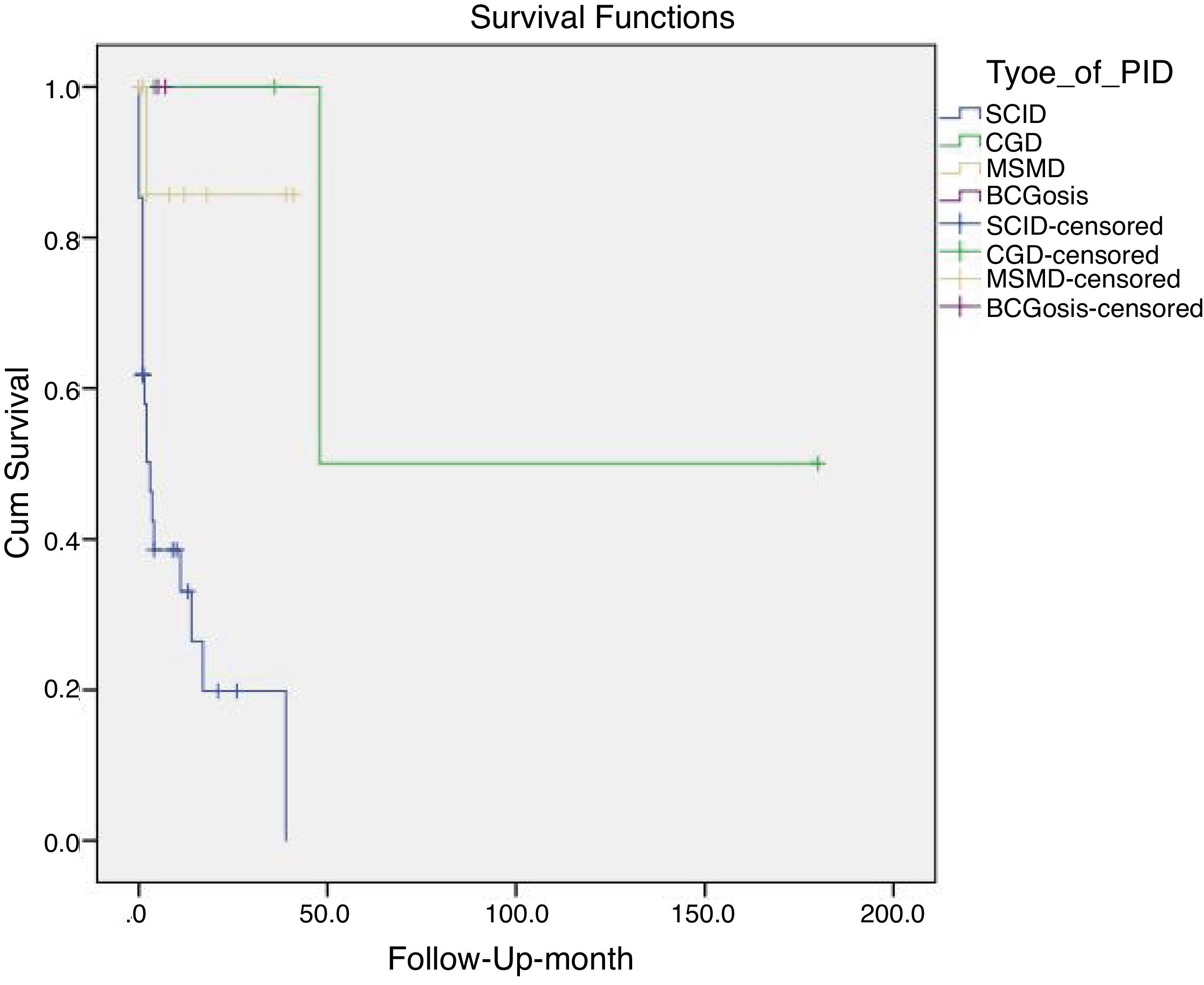
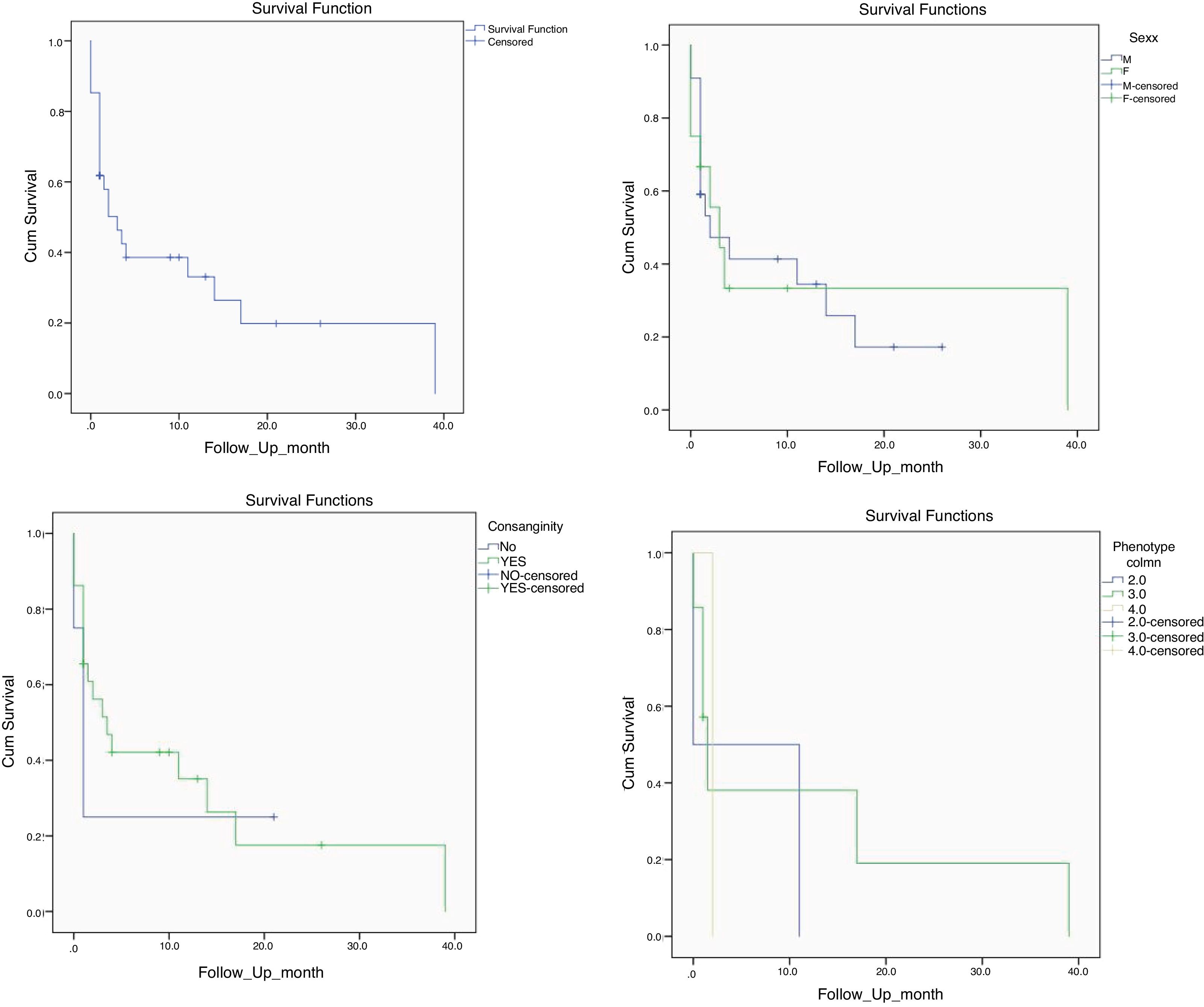
Mortality and clinical consequenceAt the time of this study, 51 patients were alive, and unfortunately, the rest have passed away. Higher rates of mortality were related to SCID patients (66.7%), as a result of a delay in diagnosis (p = 0.001). The most common cause of death was a cardio-respiratory failure due to pneumonia, systemic shock, and sepsis (68.96%). Disseminated and localized BCGosis were solely identified as the cause of death in 27.58% and 3.44% cases, respectively (Figs. 1 and 2). Moreover, the survival analysis of SCID patients with BCGosis showed the mortality rate to be higher in males and those without consanguinity. However, the mortality rate did not vary among the different SCID immune phenotypes (Fig. 3).
Discussion

 75 severe combined immunodeficiency patients and its comparison considering (B) sex, (C) parental consanguinity, and (D) phenotype (2; T-B-NK+, 3; T-B + NK-, 4; T-B + NK+).")
In this study, most of the patients registered in our national PID database with BCG complications (75 out of 137, 54.7%) were diagnosed with SCID. This data is in line with the study by Norouzi et al. that showed the most common type of immunodeficiency disorders in patients with BCGosis was SCID.7 The production and development of T cells are genetically defective in these patients and can also affect other lymphoid cells, such as B and NK cells.7 The most important known factor against BCG infections is IFN-gamma, a cytokine that is produced by T and NK cells in response to IL12/23 stimulation, which is absent or decreased in SCID patients. The impaired production of this cytokine may justify the high frequency of BCGosis in SCID patients. This study also showed that the total lymphocytes count, such as CD4+ and CD8 + T cells, and the level of immunoglobulins have increased in MSMD and CGD patients, while it has significantly decreased in SCID patients. These findings agree with the study by Marciano et al. 16, which suggested BCGosis is more frequent in SCID cases due to a low number of T cells in these patients. We also observed that T-B + NK- which was the most frequent phenotype among SCID patients (40.7%), had a more severe complication. Accordingly, BCGosis manifestations in our SCID patients showed more severity among all three groups of the patients. Compared with other cases, the incidence of pneumonia, oral candidiasis, and hepatomegaly was significantly higher in SCID patients, which put this group of patients at the highest risk of death.
In our study, 34.3% of patients with BCGosis were diagnosed with MSMD. In a single-center study from our national registry, Sadeghi-Shabestari et al. showed that among 48 selected infants with BCGosis, 11 infants had a disseminated BCG infection, and two cases had MSMD.17 In research conducted by Bousfiha et al., it was shown that five out of 12 Moroccan children with BCGosis had MSMD.18 These results confirm the susceptibility of MSMD patients to BCG infection. MSMD is a hereditary disease caused by a defect in the production or function of IL-12 and IFN-gamma loop between the T cell and the antigen-presenting cells that play an important role in the fight against Mycobacterium infection.19 The autosomal recessive inheritance is the most common genetic pattern of MSMD, and mutations in IFNGR1 and IL12Rβ1 genes are responsible for approximately 80% of all MSMD cases. Therefore, this entity of PID is higher in BCGosis cases reported from countries with a higher level of parental consanguinity.19 According to our previous genetic study from the same cohort, among 31 children with clinical signs of disseminated infections that were evaluated for MSMD, nine had mutations in IL-12Rβ1 and one had a mutation in IFNGR1.19 In another study by Ying et al., 14 out of 74 patients had a disseminated BCG infection, among which two patients had mutations in IL-12Rβ1, and two patients had mutations in the IFNGR1 gene.6 Consistent with all of the above studies, from nine of our MSMD patients in this study that were genetically evaluated, eight had mutations in IL12RB1, and one was mutated in the IFNGβ1 gene (unpublished data).
Among the 137 patients with BCGosis in this study, 15 patients (about 10%) were diagnosed with CGD, which is in line with the previous studies,6,7 suggesting that CGD patients are also susceptible to BCG complications. In patients with CGD, immune system phagocytes including macrophages, monocytes, and neutrophils, are not capable of killing and eliminating microorganisms. This inability to remove these microorganisms is due to various mutations in various genes that interfere with the production of reactive oxygen species (ROS),7 including CYBB, CYBA, NCF2, and NCF4 genes.7 In our study, among several patients that were genetically analyzed, one patient with CGD had a mutation in the CYBA gene. Ying et al. performed mutation analysis for 23 CGD patients who developed BCGosis, and 17 of the patients had mutations in the CYBB gene, two patients had mutations in NCF2, one patient had a mutation in the CYBA gene and three cases had no mutation.6 They also showed that there is no significant association between the severity of the BCG infection and the mutated genes.6 Moreover, the most frequent form of PIDs in Chinese children with BCGosis was CGD. This finding contradicts several earlier reports15 and also our study has shown that most patients with BCGosis have a diagnosis of SCID.7 This difference might be due to the lack of sufficient registration regarding SCID and the early death of children with SCID or different genetics of the population in China.6
ConclusionPIDs can lead to BCGosis with various severities. In addition to the severity of the complications, the onset of these manifestations also varies. Overall, BCGosis in patients with SCID and CGD presents more aggressive features than other immunodeficiencies like MSMD.7 Parental consanguinity is a known risk factor for the development of an autosomal recessive form of PIDs. Since the rate of consanguineous marriage in Iran is high, many children with immunodeficiency are at risk of developing BCGosis. Thus, a regular and accurate screening program (at least discovery of SCID by counting T cell receptor excision circles in neonatal Guthrie card) in addition to the proper and early diagnosis of PIDs is needed. Perhaps, paying attention to the family history of immunodeficiency disorders, especially before BCG vaccination, seems extremely necessary. Furthermore, a manifestation of BCGosis could be an important indicator of early diagnosis of PIDs. Thus, increasing awareness of physicians about the association of BCGosis and PIDs is necessary and will lead to earlier diagnosis of these heterogeneous groups of disorders.
FundingThis work was supported by a grant (no. 94-03-154-29144) from Tehran University of Medical Sciences, Tehran, Iran.
Conflict of interestThe authors declare no conflict of interest.
