





Antibodies are an essential component of the adaptative immune response and hold long-term memory of the immunological experiences throughout life. Antibody defects represent approximately half of the well-known primary immunodeficiencies requiring immunoglobulin replacement therapy. In this article, the authors review the current indications and therapeutic protocols in the Latin American environment. Immunoglobulin replacement therapy has been a safe procedure that induces dramatic positive changes in the clinical outcome of patients who carry antibody defects.
Antibodies are essential for defence against infections not only because they specifically recognise myriad microorganisms, but also for their effector capacity, which includes neutralisation, opsonisation, antibody-dependent cytotoxicity, and clearance of immune complexes.1 The importance of antibodies is clearly demonstrated by the high infection-associated morbidity and mortality in patients with primary immunodeficiency diseases (PIDD) commonly associated with impairments in antibody production.2,3 The first immunological description of PIDD was made by Ogden Bruton in 1952 in a patient who presented with recurrent infections and several episodes of sepsis. The disease was termed agammaglobulinaemia, as it was identified by the absence of the gamma fraction after immunoelectrophoresis of serum from the patient. Eventually, he was treated with human immunoglobulin and this intervention led to a decrease in the frequency of infections.4 There have been remarkable advances in the understanding of PIDD since this first report. To date, more than 180 genetic defects clustered in eight groups have been classified according to their major associated immunological defects.5 Many affected patients have an increased susceptibility to recurrent infections—in some cases with high specificity to certain microorganisms—as well as autoimmunity, chronic inflammation, and neoplasia. Approximately 50% of PIDD affect antibody-mediated immunity,6–9 and can be treated with human immunoglobulin preparations. Standard human immunoglobulins were first developed in the 1950s, while the introduction of intravenous (IVIG) and subcutaneous (SCIG) preparations in the 1980s and at the end of the 1990s brought significant advances to the field of immunoglobulin replacement therapy (IRT) in PIDD.10–12 Immunoglobulins are now employed to treat a wide range of other conditions that are associated with hypogammaglobulinaemia, including graft versus host disease, multiple myeloma, and chronic lymphoid leukaemia, as well as diseases in which their “immunomodulatory” properties have a major impact such as idiopathic thrombocytopenia purpura, Kawasaki disease, Guillain–Barré syndrome, and polymyositis/dermatomyositis.13
Appropriate treatment with IVIG or SCIG is presently the most important aspect of management for many individuals with PIDD. This intervention may be lifesaving, and can improve the quality of life for many patients. Benefits of immunoglobulin therapy are multiple and include significant decreases in the long-term cost of healthcare.14 Unfortunately; IVIG and SCIG are not prescribed for many patients requiring these treatments.
Multiple guidelines have been developed for the use of IVIG and SCIG.15–17 The Brazilian guidelines,18 written by the Grupo de Assessoria Científica sobre Imunodeficiências da Associação Brasileira de Alergia e Imunopatologia (ASBAI), were the first to be developed in Latin America and were adapted from the North American,16,17 IUIS,15 and European guidelines19 for the management of patients with PIDD. These new guidelines revise, amend, and update their recommendations for implementation across the region and include expert advice from several specialists—in particular, members of the Latin American Society for Immunodeficiencies (LASID). This document represents the consensus opinion of experts in the field regarding the use of IVIG and SCIG for the replacement of antibodies, and for prophylaxis against infections in patients with PIDD in Latin America.
ObjectivesThe objective of these guidelines is to provide recommendations and guidance regarding:
- 1.
Indications and dosing for immunoglobulin replacement therapy
- 2.
Monitoring dosing, plasma levels, and patient responses to immunoglobulin administration
- 3.
Preventing, monitoring, and managing side effects
- 4.
Monitoring patients for infections
- 5.
The latest information for available products by country
Current evidence from clinical studies supports the use of IVIG in patients with antibody deficiencies, as this intervention decreases the recurrence and severity of infections—thereby reducing hospital admissions and mortality while improving quality of life.20 In addition, it has been noted that decreasing recurrent acute pulmonary infections also reduces the risk for chronic lung disease.21 Results from a recent US survey have shown that administration of immunoglobulin to patients with PIDD is cost effective. The average cost of medical care per patient in the year before diagnosis was $138,760 (USD), as compared to $60,297 in the year after diagnosis and initiation of immunoglobulin therapy, with an average annual saving per patient of $78,166.00.14
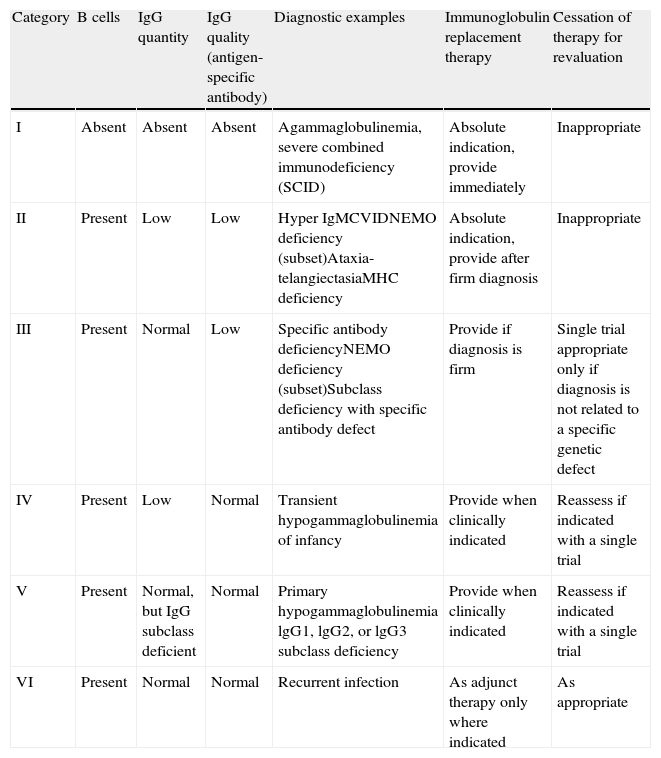
The decision to offer immunoglobulin replacement therapy should be based on: the patient's diagnosis (Table 1); proper assessment of serum immunoglobulin levels; other immunological parameters22; and on secondary causes of hypogammaglobulinaemia (Table 2). The efficacy of immunoglobulin replacement therapy in patients with certain immunodeficiencies is discussed in the following sections.
PIDD diagnoses and recommendations for immunoglobulin administration.22
| Category | B cells | IgG quantity | IgG quality (antigen-specific antibody) | Diagnostic examples | Immunoglobulin replacement therapy | Cessation of therapy for revaluation |
| I | Absent | Absent | Absent | Agammaglobulinemia, severe combined immunodeficiency (SCID) | Absolute indication, provide immediately | Inappropriate |
| II | Present | Low | Low | Hyper IgMCVIDNEMO deficiency (subset)Ataxia-telangiectasiaMHC deficiency | Absolute indication, provide after firm diagnosis | Inappropriate |
| III | Present | Normal | Low | Specific antibody deficiencyNEMO deficiency (subset)Subclass deficiency with specific antibody defect | Provide if diagnosis is firm | Single trial appropriate only if diagnosis is not related to a specific genetic defect |
| IV | Present | Low | Normal | Transient hypogammaglobulinemia of infancy | Provide when clinically indicated | Reassess if indicated with a single trial |
| V | Present | Normal, but IgG subclass deficient | Normal | Primary hypogammaglobulinemia lgG1, lgG2, or lgG3 subclass deficiency | Provide when clinically indicated | Reassess if indicated with a single trial |
| VI | Present | Normal | Normal | Recurrent infection | As adjunct therapy only where indicated | As appropriate |
Causes of hypogammaglobulinaemia other than PIDD.a
| Drugs |
| Antimalarial agents |
| Captopril |
| Carbamazepine |
| Corticosteroids |
| Fenclofenac |
| Gold salts |
| Penicillamine |
| Phenytoin |
| Sulfasalazine |
| Infectious diseases |
| Human immunodeficiency |
| Congenital rubella |
| Congenital cytomegalovirus |
| Congenital toxoplasmosis |
| Epstein–Barr mononucleosis |
| Systemic diseases |
| Immunodeficiency caused by immunoglobulin hypercatabolism |
| Immunodeficiency caused by excessive loss of immunoglobulins (nephrosis, extensive burn, lymphangiectasia, diarrhoea, enteropathy with loss of proteins) |
| Neoplasms |
| Chronic lymphocytic leukaemia |
| Non-Hodgkin's lymphoma |
| B cell neoplasy |
These patients commonly have <2% of the normal levels of B lymphocytes in peripheral blood along with severe reduction in all serum Ig isotypes. The use of IVIG in these patients has been shown to decrease the risk for serious infections.23,24 Retrospective analyses of children with agammaglobulinaemia have shown that the frequency and severity of infections are inversely proportional to the dose of IVIG and that serum concentrations of IgG >500mg/dL are effective for the prevention of severe bacterial infections—including pulmonary infections and meningoencephalitis—thus directly decreasing mortality and improving quality of life.24,25
Common variable immunodeficiency and other primary hypogammaglobulinaemias: severe reduction in at least two serum immunoglobulin isotypes with normal or low number of B cellsThese conditions are characterised by a reduction of serum IgG of at least two standard deviations below the mean for patient age, and in the case of CVID at least one other serum immunoglobulin isotype (often IgM or IgA) with normal or low numbers of B cells. These patients are at increased risk for recurrent infections of the respiratory tract and chronic pulmonary disease.26,27 Treatment with IVIG has been shown to significantly decrease the susceptibility to recurrent infections in patients with common variable immunodeficiency (CVID) and other primary hypogammaglobulinaemias.26 Early identification of these patients and treatment with IVIG are crucial for reducing the occurrence of pneumonia and the progression of chronic lung disease.28,29
Hyper-IgM syndrome or class-switch recombination defects: severe reduction in serum IgG and IgA with normal/elevated IgM and normal numbers of B cellsClass-switch recombination defects are characterised by reduced levels of serum IgG and IgA, along with normal-to-high levels of serum IgM, and limited production of specific antibodies after exposure to T cell-dependent antigens. The number of B lymphocytes is normal, but patients present with recurrent infections clinically similar to those in patients with agammaglobulinaemia as well as fungal and other intracellular pathogens. Treatment with IVIG decreases infections in these patients.30,31
Specific antibody deficiency with normal immunoglobulin levels and normal numbers of B cellsTreatment with IVIG can be indicated in patients with recurrent infections of the upper and lower respiratory tract, and this treatment can also be included and indicated as a documented requirement for antibiotic therapy for patients who exhibit normal serum immunoglobulin levels along with a well-documented lack of response to polysaccharide antigens (most frequently vaccinal antigens).32
IgA deficiency associated with IgG subclass deficienciesThere are no commercially available preparations of enriched IgA or IgM, so patients who have isolated IgA deficiency generally are not considered candidates for IgG supplementation, except when there is an association with IgG subclass deficiencies and significant morbidity.33 In these patients, IVIG or SCIG preparations with low concentrations of IgA must be used because of the possibility of increased risk of adverse events.21,34,35
Transient hypogammaglobulinaemia of infancyThis condition is the most common cause of symptomatic hypogammaglobulinaemia in children under the age of two years. The diagnosis can only be confirmed when serum immunoglobulins reach age-appropriate levels. In general, the disease has a benign course, but some children may develop serious infections. Therefore, treatment with IVIG should be considered in some cases as it has been shown to reduce the severity of infections in these children.36,37
Combined immunodeficienciesCombined immunodeficiencies or defects in T lymphocyte-mediated immunity are the most severe forms of PIDD. In some types of severe combined immunodeficiency (SCID), B cells and natural killer (NK) cells may be present but have inadequate functionality.15 SCID is invariably fatal and the only curative treatment that reduces mortality in these patients is haematopoietic stem cell transplantation (HSCT) at an early age. Administration of IVIG is indicated immediately after diagnosis and should be maintained during and after HSCT, as most patients exhibit persistently impaired B-cell function despite reconstitution of T cells.38,39 If chimerism produces T cells from a donor and B cells from a recipient, the patient will probably need long-term IgG replacement with IVIG.38
Well-defined syndromes with immunodeficiencyHyper IgE syndrome (HIES)Patients with HIES due to STAT3 mutations usually exhibit very high serum IgE but present normal levels of serum IgG, IgM, and normal or low serum IgA; some also have deficient antibody production of T-independent antigens such as Pneumococcal and Haemophilus influenzae.40 In contrast, patients with HIES due to DOCK-8 mutations often have high serum IgG and almost all have high IgE as well—although IgM is usually low, while IgA can be high or low.41 Patients with hyper IgE syndrome who develop respiratory infections might benefit from IVIG administration.42 The current understanding is that IVIG should only be used in patients for whom specific antibody deficiency is demonstrated.43
Wiskott–Aldrich syndromeThe humoral abnormalities in Wiskott–Aldrich syndrome are related to the production of specific antibodies against proteins and polysaccharides. Although serum IgG is relatively normal, serum IgM can be low while serum IgA and IgE are elevated. Administration of IVIG reduces the risk of infections and must be administered while the patient's immune system is being reconstituted by haematopoietic stem cell transplantation.44,45
Ataxia-telangiectasiaAbout 70% of the patients with ataxia-telangiectasia exhibit IgA deficiency, and some also have IgG subclass deficiencies and defective antibody production against polysaccharides. These patients may benefit from IVIG treatment,46,47 particularly if they have IgG2 deficiency or agammaglobulinaemia.
WHIM syndromeIn patients with warts, hypogammaglobulinaemia, infections, and myelokathexis (WHIM) syndrome, the administration of IVIG has been shown to decrease the number and severity of infections.48
Bone marrow transplantationTreatment of combined immunodeficiency requires reconstitution by haematopoietic stem cells. Bone marrow transplantation has been applied successfully in cellular immunodeficiencies, severe combined immunodeficiency, and many other combined immunodeficiencies.32 Administration of IVIG is indicated as part of the support regimen for these patients both before and after transplantation.49
In conclusion, the principal indications for immunoglobulin replacement therapy have been presented. Any additional indications will result when doctors find patients with recurrent infections, IgG levels below 200mg/dL or lack of specific antibodies.
Guidelines for the use of IVIG in PIDDAmerican Academy of Allergy, Asthma and ImmunologyIVIG is indicated as replacement therapy for PIDD patients with defective antibody production. The recommendations for its indications, according to the Primary Immunodeficiencies Committee of the American Academy of Allergy, Asthma and Immunology20 are as follows:
Definitively beneficial- •
Primary immune defects with absent B cells
- •
Primary defects with hypogammaglobulinaemia and impaired specific antibody production
- •
Primary immune defects with normogammaglobulinaemia and impaired specific antibody production
- •
Isolated IgA deficiency1
- •
Isolated IgG4 deficiency
The International Union of Immunological Societies has stated that all patients with primary specific immunodeficiency who have significantly diminished serum immunoglobulin levels and/or demonstrated defects in antibody production should receive immunoglobulin replacement therapy. They note that replacement therapy with IVIG can save lives and, if started early with appropriate dosing, can arrest the cycle of recurrent infections and progressive lung damage that occurs in many patients with PIDD.50
European Society for ImmunodeficienciesThe European Society for Immunodeficiencies (ESID) recommends the following approach to determine when the administration of IVIG is indicated for reconstitution of serum immunoglobulins19:
- •
IgG <200mg/dL: all patients (excluding children that may present with physiologic hypogammaglobulinaemia without severe infections)
- •
IgG levels 200–500mg/dL: when the deficiency is identified and associated with recurrent infections
- •
IgG >500mg/dL: when there is a deficiency in the formation of antibodies to specific antigens and serious or recurrent infections
Although the most important reason for the administration of immunoglobulins is the prevention of pneumonia and other serious infections, additional parameters such as pulmonary function and quality of life should be evaluated in order to provide a more complete assessment of the patient's status.51 This section summarises pre-treatment of patients with PIDD who are candidates for immunoglobulin replacement therapy, dosing for IVIG, and important aspects of patient assessment and monitoring.
Pre-treatment assessmentsBefore the initiation of immunoglobulin therapy, physicians must carry out a detailed clinical evaluation, including laboratory tests, to provide a comprehensive assessment of the patient's health condition.21,52 Such evaluation should include:
- •
Serum levels of all immunoglobulins (IgG, IgA, IgM, and IgE)
- •
Titres of antibodies specific to protein and polysaccharide antigens from vaccines (may not be necessary except in cases of specific antibody deficiencies, since levels <200mg/dL might give false negative titres)
- •
Complete blood count/blood smear
- •
Lymphocyte populations in peripheral blood (B, T, and NK lymphocytes) and in some cases specific subsets, such as memory B and T cells or other subpopulations, when available
- •
Evaluation of organ function, especially in patients at increased risk for sequelae (e.g., evaluations of the lungs should ideally include pulmonary function testing and high-resolution contrasted computed tomography)
- •
Assessment of hepatic and renal function (total and direct bilirubin, transaminases, gamma glutamyl transferase, lactate dehydrogenase, blood urea nitrogen, and serum creatinine)
- •
Assessment of infectious diseases with possible blood transmission such as HIV, hepatitis B, hepatitis C, toxoplasmosis, cytomegalovirus infection, mononucleosis, rubella, Chagas disease, malaria, leishmaniasis, herpes simplex virus infection, or other infectious diseases according to the geographical particularities
- •
Definition of the genetic defect, if feasible
- •
Collection of a 5–10mL serum and/or plasma sample (with EDTA) prior to initiation of treatment with IVIG and storage at −80°C for future analysis if necessary.
Current dosing for IVIG in patients with PIDD is based on results from clinical trials focused on the prevention of infections, but another objective is to prevent the long-term consequences of recurrent infections to target organs such as the lungs. Although this goal is important, clinical results to date have not provided guidance regarding the dosing necessary to achieve it. Therefore, patients should be constantly monitored for the development of complications or sequelae, particularly in the lungs, gastrointestinal tract, and central nervous system.28 Results from a recent meta-analysis show the importance of a treatment aimed at achieving the desired clinical outcome, as opposed to achieving only a desired plasma IgG level.53 This study combined results from all available studies evaluating trough immunoglobulin levels and pneumonia incidence in PIDD patients with hypogammaglobulinaemia who received IVIG. It included results from 17 studies and 676 patients (2127 patient-years of follow-up). These results indicated that the incidence of pneumonia declined by 27% with each 100mg/dL increment in trough IgG levels. Pneumonia incidence with maintenance trough levels of 500mg/dL was five times more frequent than with a trough level of 1000mg/dL (0.113 versus 0.023 cases per patient-year).53
In general, the recommended initial dose of IVIG for patients with hypogammaglobulinaemia is 400–600mg/kg, although it may vary depending on the severity of the hypogammaglobulinaemia and infections. Although patients should receive IVIG infusions every 3–4 weeks to maintain IgG levels adequate to prevent severe infections,54,55 it is necessary to remember that most IVIG will initially redistribute in the tissues. By the end of an infusion, most IgG administered is found in the intravascular compartment; its concentration increases 100–200mg/dL for every 100mg/kg, reaching levels of more than 1000mg/dL after a dose of 300–800mg/kg. About 48–72h after the infusion, the serum IgG is distributed to the interstitial compartment and the serum concentration is reduced by about 25–40%. The half-life for IgG is about 22 days, justifying a dosing interval of 3–4 weeks.33 As a result, serum levels may decrease before the next scheduled dose. Bonagura et al. have suggested that the goal of IgG replacement should be to identify and maintain the biologic IgG level of an individual patient with PID within the age-matched control range, and they have coined the term “biologic IgG level” to represent the minimal serum IgG level that renders patients as disease-free as possible.56
It is also important to note that the metabolism of IgG varies among patients and is influenced by genetic factors as well as each patient's clinical status, including the presence of infections, endocrine disturbances, and autoimmunity—all of which may increase its metabolism.36 Nevertheless, in most patients, IgG levels become stable after the sixth infusion; at that time, dosing and intervals of administration may be reevaluated to achieve the best clinical response.20,21 In addition, monitoring of PIDD patients who require IgG replacement must be individualised (although a general rule is every 3–6 months, depending on the clinical response, particularly at the beginning of the treatment). “Biologic” IgG level is likely to be unique for each patient, always in the range of age-matched control subjects, and should be considered to be a moving target that can change with comorbid conditions/diseases. We recommend charting clinical infections against IgG levels over time to identify and maintain each patient's biologic IgG level, help optimise care, and address IgG reimbursement queries should they arise.56
Considerations for special patient groupsPatients with serum IgG levels <100mg/dL may benefit from doses of up to 800mg/kg, split into two infusions a few days apart, followed by monthly infusions at regular doses.36 It has been suggested that patients with IgG levels <100mg/dL at diagnosis should have trough levels maintained at >600mg/dL, and that those with a basal level of 300mg/dL and no functional antibodies should have a trough target level of 900mg/dL.57
Some patients, notably those with recurrent pulmonary infections, may require higher IVIG doses (>700mg/kg/month). IVIG doses of up to 800mg/kg have been shown to improve lung function and have been prescribed for patients with chronic lung disease, granulomatous disease, interstitial lung disease, obliterative bronchiolitis, and chronic sinusitis.58–61 This guidance is consistent with current practice in Argentina and Colombia as well. At the Children's Hospital Ricardo Gutiérrez, and other immunology centres in Argentina and Colombia, a serum concentration of 700mg/dL is used as the target for treatment because it is more effective for prevention of bacterial infections than lower target levels. A target of 800mg/dL is also considered useful for prevention of meningoencephalitis.62,63
It has been suggested that the occurrence of three or more mild infections per year justifies increasing the dose of IVIG by 150mg/kg/month or reducing the dosing interval.64 Patients with recurrent pulmonary infections and impaired lung function may also require prophylactic treatment with antimicrobial drugs, bronchodilators, short courses of corticosteroids, and/or mucolytics, as well as physical therapy.
A recent study by Lucas et al. evaluated the efficacy of IVIG in patients with X-linked agammaglobulinaemia and CVID as these patients were followed for 22 years.64 It was observed that most infections occurring while patients were under treatment involved the respiratory tract and were associated with encapsulated bacteria. Also, in contrast to patients with CVID, those with X-linked agammaglobulinaemia required higher IVIG doses to prevent infections. The study concluded that the aim of IVIG treatment in patients with antibody deficiencies should be to minimise infections, and that dosage determination should be based not only on the level of serum IgG reached, but also on clinical results.64 This view is strongly supported by the meta-analysis carried out by Orange et al.53
Monitoring patients for adverse eventsThere is no need for special equipment to monitor patients receiving IVIG, but they should be instructed to report any adverse events (AEs). Whereas most AEs associated with IVIG infusion are mild, a qualified healthcare professional should be available to manage any adverse reactions.65,66 According to the Immune Deficiency Foundation (IDF), 44% of patients receiving IVIG experience AEs during infusion. During the first infusion, 30% have side effects that either decrease in severity or do not recur with subsequent treatments, as long as the same product is used.67
Adverse events associated with IVIGThe most common side effects of IVIG treatment, recorded from patients receiving a total of over 3000 infusions, are summarised in Fig. 1.66 The most common infusion-related adverse events are chills and fever (about 3% and 1% of infusions, respectively). The frequency of AEs is increased in the presence of infections and with the first infusion.68–70 Many AEs associated with the administration of IVIG are believed to be related to the formation of immune complexes and occur less often when patients are previously and effectively treated for infections. Immune complexes also may form with lyophilised products that require dilution prior to use. To minimise immune complex formation, proper reconstitution of these products with adequate diluent requires particular attention to factors such as temperature. It has also been noted that frequent switching of products for immunoglobulin reconstitution71–73 and rapid infusion both increase the risk for AEs. Adverse events may also be related to variations in the properties of different IVIG preparations, including sugar content—particularly sucrose—hyperosmolarity, and IgA (Table 3).

Adverse events associated with 3004 infusions in patients with PIDD.66
More serious, rare AEs associated with IVIG infusion include aseptic meningitis, renal insufficiency, thrombosis, haemolytic anaemia, rashes, and anaphylaxis.74,75 Renal insufficiency is associated with the use of products containing sucrose.76 Migraines may occur up to 48–72h after infusion.33,75 Haemolytic anaemia is very rare, and patients should be monitored periodically with a direct Coombs test.77 It should be noted that most serious AEs occur during the first 30min of the infusion.73
Avoiding and managing adverse eventsPrior to the initiation of treatment with IVIG, the physician must be aware of any factors that may increase the risk for AEs, such as use of oral contraceptives, smoking, infections, diarrhoea with dehydration, and hypercoagulability states, among others.33 In Brazil, the infusions should be administered at a qualified infusion centre or day hospital. In Chile, however, IVIG cannot be administered at home or in smaller centres and is only infused in major public or private hospitals in major cities of each region of the country. In Argentina, SCIG has been in use since 2010, with good acceptance by patients. In Mexico, only IVIG is available through the government health security system, and the vast majority of infusions are done at large referral hospitals. SCIG is commercially available for private patients but it is still not included in the “basic drug catalogue” from the government health system. In Colombia, healthcare providers have developed state-of-the-art special infusion centres where patients receive IVIG. However, since the introduction of SCIG, patients receive their first few treatments at the centres and then begin a nurse-monitored programme of home infusions which continues until the patient is “certified” to perform the procedure by himself.
Several studies have analysed the efficacy of IVIG from different manufacturers; the results have been very similar, with no significant efficacy differences among products.21 However, some patients may experience more frequent AEs with some products than with others. In addition, some patients have strong reactions to products with high IgA concentrations; for these individuals, the use of low-IgA preparations must be considered. Thus, AEs specific to one product or another justify switching products or changing the administration route.21 Factors known to increase the risk for AEs in patients being infused with IVIG include:
- •
High infusion rates
- •
First-ever infusion or first infusion after a long period without treatment
- •
Acute bacterial infection
- •
Switching IVIG preparations
- •
Presence of IgA in the preparation
- •
Hyperosmolarity
The following precautions should be taken to minimise the risk of complications during and shortly after IVIG infusion:
- •
Observe the patient for AEs for ≥20min after the infusion.
- •
Monitor for symptoms suggestive of adverse events during the infusion, especially when the product is changed or when there has been a long interval between infusions.
- •
Do not use preparations containing a sugar stabiliser in patients with diabetes.
- •
Carefully assess risk factors for kidney failure, including pre-existing renal insufficiency, diabetes mellitus, hypovolaemia, dehydration, obesity, use of nephrotoxic drugs, and age >65 years.
In addition, AEs can be reduced by initiating infusions at a slow rate of 0.01mL/kg/min (equivalent to 0.5mg/kg/min for a 5% solution or 1mg/kg/min for a 10% solution) using an infusion pump. Vital signs should be monitored at regular intervals; if no AEs are noted, the infusion rate may be increased every 30min, up to a maximum of 0.08mL/kg/min (equivalent to 4mg/kg/min for a 5% solution or 8mg/kg/min for a 10% solution).33 For example, in a 50-kg patient, a 500mg/kg/dose=25g, and the total infusion volume for a 5% concentration solution is 500mL. An initial infusion rate of 0.01mL/kg/min corresponds to 30mL/h. If there are no AEs after the first 30min, the infusion rate may be increased at defined intervals up to 0.08mL/kg/min, which corresponds to 240mL/h. The infusion will take about 3h, assuming that the patient does not develop an AE. If a 10% solution is used, the infusion time may be reduced by 50%. Therefore, infusion rates vary with the product used, and the manufacturer's instructions must be followed at all times (Table 4). Patients should be strongly encouraged to hydrate themselves; drinking water should be available and easily accessible at all times.
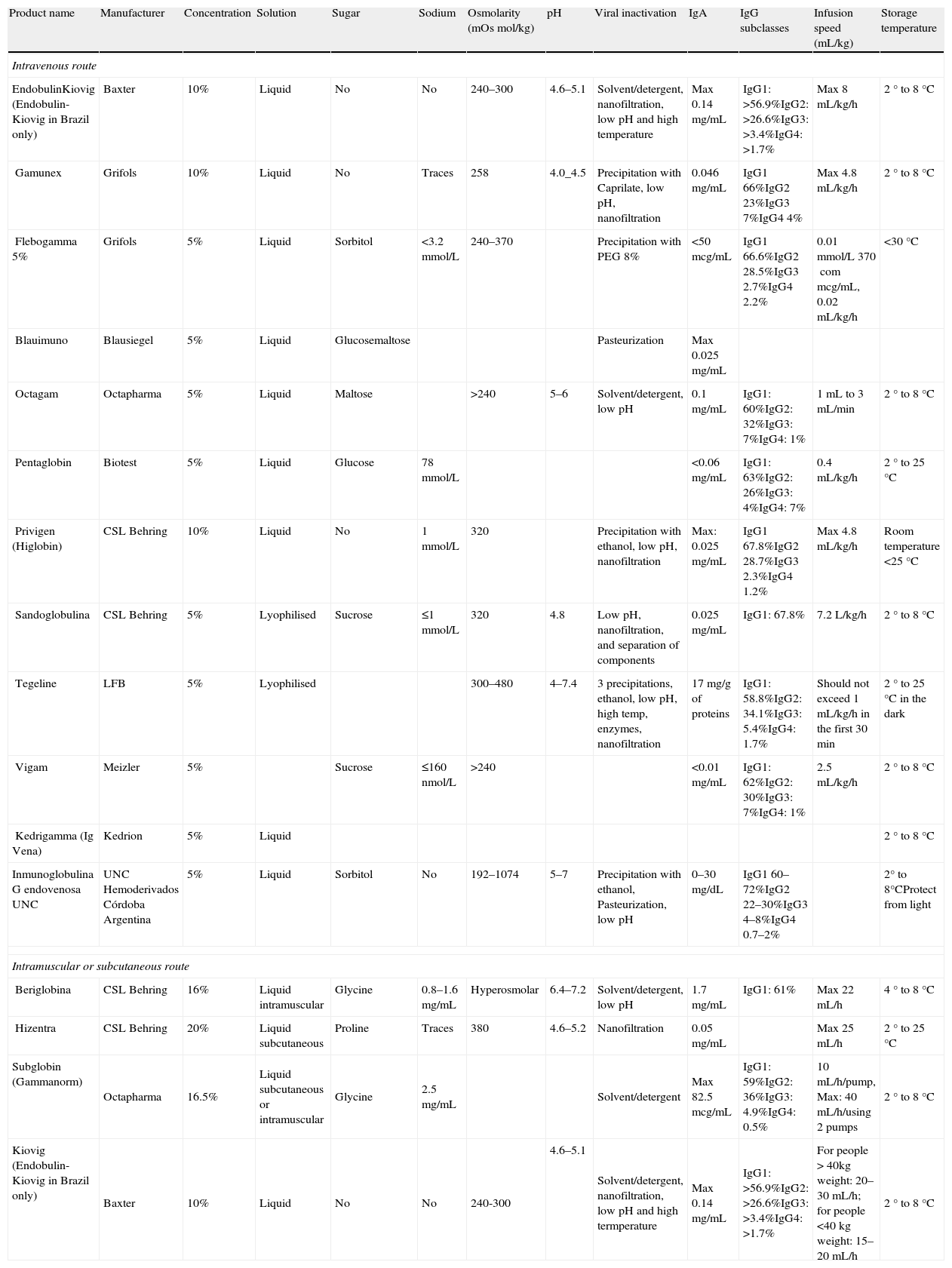
Currently available IVIG preparations.
| Product name | Manufacturer | Concentration | Solution | Sugar | Sodium | Osmolarity (mOsmol/kg) | pH | Viral inactivation | IgA | IgG subclasses | Infusion speed (mL/kg) | Storage temperature |
| Intravenous route | ||||||||||||
| EndobulinKiovig (Endobulin-Kiovig in Brazil only) | Baxter | 10% | Liquid | No | No | 240–300 | 4.6–5.1 | Solvent/detergent, nanofiltration, low pH and high temperature | Max 0.14mg/mL | IgG1: >56.9%IgG2: >26.6%IgG3: >3.4%IgG4: >1.7% | Max 8mL/kg/h | 2° to 8°C |
| Gamunex | Grifols | 10% | Liquid | No | Traces | 258 | 4.0_4.5 | Precipitation with Caprilate, low pH, nanofiltration | 0.046mg/mL | IgG1 66%IgG2 23%IgG3 7%IgG4 4% | Max 4.8mL/kg/h | 2° to 8°C |
| Flebogamma 5% | Grifols | 5% | Liquid | Sorbitol | <3.2mmol/L | 240–370 | Precipitation with PEG 8% | <50mcg/mL | IgG1 66.6%IgG2 28.5%IgG3 2.7%IgG4 2.2% | 0.01mmol/L 370commcg/mL, 0.02mL/kg/h | <30°C | |
| Blauimuno | Blausiegel | 5% | Liquid | Glucosemaltose | Pasteurization | Max 0.025mg/mL | ||||||
| Octagam | Octapharma | 5% | Liquid | Maltose | >240 | 5–6 | Solvent/detergent, low pH | 0.1mg/mL | IgG1: 60%IgG2: 32%IgG3: 7%IgG4: 1% | 1mL to 3mL/min | 2° to 8°C | |
| Pentaglobin | Biotest | 5% | Liquid | Glucose | 78mmol/L | <0.06mg/mL | IgG1: 63%IgG2: 26%IgG3: 4%IgG4: 7% | 0.4mL/kg/h | 2° to 25°C | |||
| Privigen (Higlobin) | CSL Behring | 10% | Liquid | No | 1mmol/L | 320 | Precipitation with ethanol, low pH, nanofiltration | Max: 0.025mg/mL | IgG1 67.8%IgG2 28.7%IgG3 2.3%IgG4 1.2% | Max 4.8mL/kg/h | Room temperature <25°C | |
| Sandoglobulina | CSL Behring | 5% | Lyophilised | Sucrose | ≤1mmol/L | 320 | 4.8 | Low pH, nanofiltration, and separation of components | 0.025mg/mL | IgG1: 67.8% | 7.2L/kg/h | 2° to 8°C |
| Tegeline | LFB | 5% | Lyophilised | 300–480 | 4–7.4 | 3 precipitations, ethanol, low pH, high temp, enzymes, nanofiltration | 17mg/g of proteins | IgG1: 58.8%IgG2: 34.1%IgG3: 5.4%IgG4: 1.7% | Should not exceed 1mL/kg/h in the first 30min | 2° to 25°C in the dark | ||
| Vigam | Meizler | 5% | Sucrose | ≤160nmol/L | >240 | <0.01mg/mL | IgG1: 62%IgG2: 30%IgG3: 7%IgG4: 1% | 2.5mL/kg/h | 2° to 8°C | |||
| Kedrigamma (Ig Vena) | Kedrion | 5% | Liquid | 2° to 8°C | ||||||||
| Inmunoglobulina G endovenosa UNC | UNC Hemoderivados Córdoba Argentina | 5% | Liquid | Sorbitol | No | 192–1074 | 5–7 | Precipitation with ethanol, Pasteurization, low pH | 0–30mg/dL | IgG1 60–72%IgG2 22–30%IgG3 4–8%IgG4 0.7–2% | 2° to 8°CProtect from light | |
| Intramuscular or subcutaneous route | ||||||||||||
| Beriglobina | CSL Behring | 16% | Liquid intramuscular | Glycine | 0.8–1.6mg/mL | Hyperosmolar | 6.4–7.2 | Solvent/detergent, low pH | 1.7mg/mL | IgG1: 61% | Max 22mL/h | 4° to 8°C |
| Hizentra | CSL Behring | 20% | Liquid subcutaneous | Proline | Traces | 380 | 4.6–5.2 | Nanofiltration | 0.05mg/mL | Max 25mL/h | 2° to 25°C | |
| Subglobin (Gammanorm) | Octapharma | 16.5% | Liquid subcutaneous or intramuscular | Glycine | 2.5mg/mL | Solvent/detergent | Max 82.5mcg/mL | IgG1: 59%IgG2: 36%IgG3: 4.9%IgG4: 0.5% | 10mL/h/pump, Max: 40mL/h/using 2 pumps | 2° to 8°C | ||
| Kiovig (Endobulin-Kiovig in Brazil only) | Baxter | 10% | Liquid | No | No | 240-300 | 4.6–5.1 | Solvent/detergent, nanofiltration, low pH and high termperature | Max 0.14mg/mL | IgG1: >56.9%IgG2: >26.6%IgG3: >3.4%IgG4: >1.7% | For people > 40kg weight: 20–30mL/h; for people <40kg weight: 15–20mL/h | 2° to 8°C |
Some of the AEs associated with IVIG infusions, such as chills and fever, can mimic infection. Other symptoms commonly associated with IVIG infusions include arthralgia, myalgia, abdominal pain, nausea, and migraines.
The risk for infusion-associated adverse events can be reduced by administration of appropriate prophylaxis before IVIG infusion. Patients should be assessed for premedication requirements (e.g., asked whether any adverse events occurred after the previous infusion), and appropriate medications such as IV corticosteroids (hydrocortisone 10mg/kg), IV or oral antihistamines (diphenhydramine 1mg/kg), acetaminophen (15mg/kg) and/or non-steroidal anti-inflammatory drugs may be administered prior to infusion or during AEs.78 A well-hydrated status before IVIG infusion is also important to prevent AEs.
If AEs occur, the infusion must be stopped for 15–30min and the patient hydrated and treated as needed with analgesics, antihistaminics, and antiemetic drugs. After the patient is stabilised, the infusion may be resumed at the initial rate and can be increased according to the patient's response.
Since IVIG is a haemoderivative, physicians must register the name and lot number of the product being infused.33 Adverse events requiring the use of medication should be reported to the following addresses for each country.
- •
In Brazil, AEs are reported online at: http://portal.anvisa.gov.br/wps/portal/anvisa/posuso/farmacovigilancia; http://www.cvs.saude.sp.gov.br/eventos_adv.asp; and http://www.anvisa.gov.br/servicos/form/farmaco/index.htm
- •
In Chile, AE reports go to the pharmacy of each hospital. A special report form for adverse reactions to drugs is completed and then forwarded to the Instituto de Salud Pública de Chile (http://www.ispch.cl).
- •
In Mexico, AE reports go to the Comisión para la prevención de riesgos sanitarios, Secretaria de Salud, México.
- •
In Argentina, most patients receive treatment in hospitals or private clinics, and adverse events are reported to the pharmacy of each institution. The pharmacies then send AE reports to the Ministerio de Salud, Secretaría de Políticas, Regulación y Relaciones Sanitarias A.N.M.A.T. (http://www.anmat.gov.ar).
- •
In Colombia, the Ministry of Social Protection through the National Institute for the Surveillance of Medicines and Food (Instituto Nacional de Vigilancia de Medicamentos y Alimentos, INVIMA), has a national surveillance programme named “Programa Nacional de Farmacovigilancia” (PNFV). This programme is in charge of receiving the information and then reporting adverse events, evaluating risks/benefits, and communicating the results and recommendations to the medical community and patients. Awareness campaigns are carried out through the National Network of Pharmacosurveillance (Red Nacional de Farmacovigilancia), a national network of regional nodes that gathers the information from healthcare providers, physicians and patients, and works in collaboration with the pharmaceutical industry. If a report is generated, the INVIMA notifies the national network and international organisations such as the World Health Organization (WHO). In addition, Colombia is a member of the Pan-American Network for the harmonisation and regulation of pharmacologic products (Red Panamericana para la Armonización y Reglamentación Farmacéutica [PARF]). To report an adverse event, a special format called FORAM is available for download at the INVIMA official website (www.invima.gov.co).
- •
In Costa Rica, AEs are reported using the formulary “Notificación de sospecha de reacción adversa a un medicamento” and are sent to the “Centro Nacional de Farmacovigilancia, Dirección de Vigilancia de la Salud, Ministerio de Salud.”
Laboratory tests such as blood counts/blood smears erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), and polymerase chain reaction (PCR) are used to detect subclinical infections. It is also important to assess plasma immunoglobulin levels, as well as renal and hepatic function, every 6–12 months.61
Product quality and safetyThe increasing availability of IVIG and SCIG products provided by different companies poses an additional challenge. The way gammaglobulin is produced varies from one place to another and quality and effectiveness vary. Governments of countries that do not have the capability of assessing gammaglobulin products should consider which products are approved for consumption by the FDA, the European regulatory agencies, or by Latin American countries which have strict regulatory agencies.
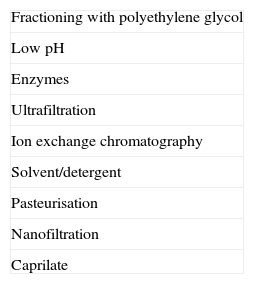
Product manufacture and regulationIn order to obtain the desired efficacy, IVIG preparations must have a high diversity of antibodies against many pathogens. Bioequivalence assessments of IVIG have not been carried out.16,20,68,73,79 These preparations are derived from human plasma. Preparation involves: selecting donors, obtaining the plasma, quarantining, and plasma fractioning; purifying products; stabilising, inactivating, and/or removing viruses and other pathogens; and preparing the final composition for clinical use (Tables 3 and 5).73
There are several IVIG manufacturers worldwide. In Brazil, IVIG products must be evaluated for efficacy and safety by the Ministério da Saúde do Brasil and ANVISA (Agência Nacional de Vigilância Sanitária). In Chile, this is accomplished by the Instituto de Salud Pública de Chile. In Mexico, IVIG products must be evaluated for efficacy and safety by the Comisión Federal para la Protección contra Riesgos Sanitarios, Secretaría de Salud (http://www.cofepris.gob.mx). In Argentina, IVIG products must be evaluated for efficacy and safely by the Ministerio de Salud, Secretaría de Políticas, Regulación y Relaciones Sanitarias A.N.M.A.T. (http://www.anmat.gov.ar). In Colombia, it is the Instituto Nacional de Vigilancia de Medicamentos y Alimentos—INVIMA (http://www.invima.gov.co). In Costa Rica, IVIG products are evaluated for registration at the Ministry of Health, and are later evaluated at the quality control labs of the Social Security System. Few studies are available that compare the efficiency and tolerability of these different products.36 The characteristics of all commercially available IVIG preparations are listed in Table 4.73,80
Transmission of infection and risk for malignancyBecause IVIG is a plasma derivative, there is always a concern about the transmission of microorganisms from donors. There is also some risk for the transmission of infection from staff at the infusion centre. In general, viral surface proteins are sensitive to low pH, proteolytic enzymes, and heating; these treatments are often employed for viral inactivation. Enveloped viruses require alternative inactivation protocols using fatty acids, alcohol, and combinations of solvents/detergents.81 Some products are further nanofiltered to remove viral particles by size, regardless of their chemical properties (Table 5).33 All of these methods are effective for the removal of viral particles. The combination of techniques used for removing viruses from commercial preparations, along with careful donor screening, have decreased the risk for viral infection to near zero. However, there are viruses that cannot be detected and other pathogens may be transmitted through gammaglobulin preparations. Patients, healthcare providers, and authorities must be aware of the risks associated with these products and all the processes that can be employed to minimise the possibility of transmission of infection.82
The risk for neoplasia in patients with PIDD is well known; however, there is no evidence suggesting that the use of IVIG increases the risk for the development of tumours.21 There is an increased risk for autoimmunity in patients with PIDD, and this is not prevented by treatment with IVIG.83,84
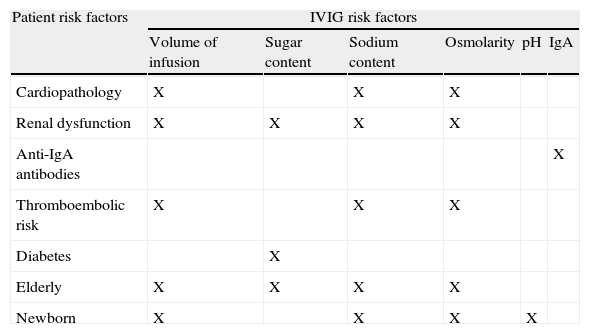
Considerations for special patient populationsIn the plasma, immunoglobulin antibodies circulate as monomers and when aggregated may give rise to AEs such as anaphylaxis and renal insufficiency. IVIG products contain excipients, such as amino acids or sugars, which minimise the formation of aggregates and maintain the immunoglobulin molecules in their monomeric form. Some of these vehicles should be avoided in certain patients. For example, products with sucrose must be avoided in patients with diabetes and/or other risk factors for renal disease.85
Patients with PIDD frequently present with comorbidities that have important clinical effects; particular care must be exercised in the treatment of those patients who may have impaired renal, cardiac, or liver function (Table 6). Products with proline should be avoided in patients with defects in amino acid metabolism, and products with sorbitol should be avoided in patients with diabetes or fructose intolerance.86 It should also be noted that blood glucose monitors detect maltose, icodextrine, galactose, and xylose as glucose; leading to erroneously high readings in patients infused with products containing any of these sugars. In addition, hyperosmolar products have a higher risk for thromboembolic events, especially in elderly patients, newborns, and those with either cardiac or renal disease.87 The high osmolarity of these preparations also increases the probability of renal AEs.54
Factors associated with increased risk for AEs with IVIG.a
| Patient risk factors | IVIG risk factors | |||||
| Volume of infusion | Sugar content | Sodium content | Osmolarity | pH | IgA | |
| Cardiopathology | X | X | X | |||
| Renal dysfunction | X | X | X | X | ||
| Anti-IgA antibodies | X | |||||
| Thromboembolic risk | X | X | X | |||
| Diabetes | X | |||||
| Elderly | X | X | X | X | ||
| Newborn | X | X | X | X | ||
Treatment with IVIG may be associated with renal AEs; the risk for such events is increased in patients with pre-existing kidney dysfunction, diabetes mellitus, age >65 years, dehydration, septicaemia, and paraproteinaemia, as well as in those taking nephrotoxic drugs. IVIG should be infused slowly in these patients, and the product used should have the lowest possible levels of sugar.82,88–92
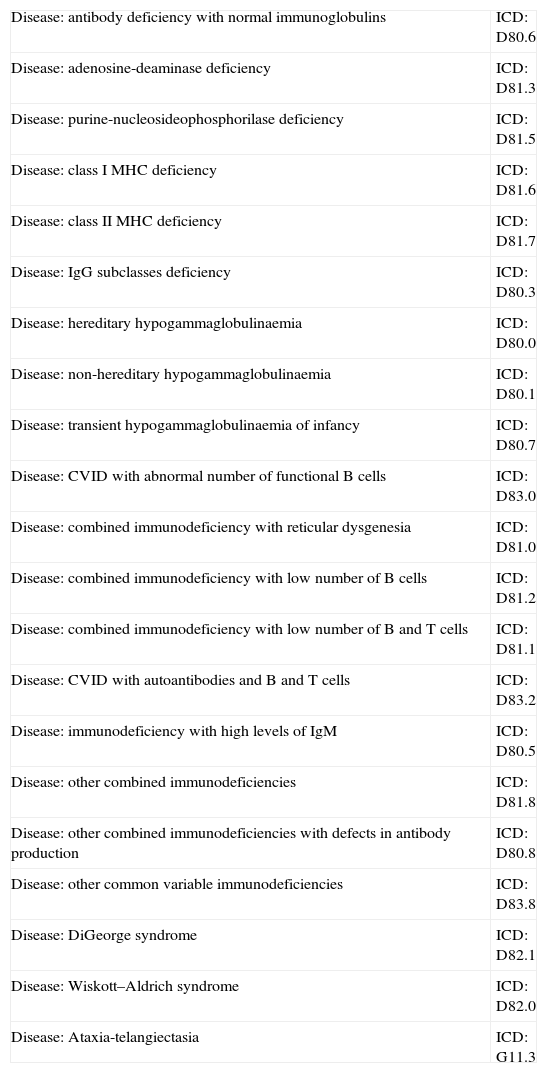
Obtaining IVIGAs soon as immunological evidence of an antibody deficiency is found in the patient (there is no need for genetic diagnosis) and the treatment is prescribed, information should be submitted to the group that provides medications for the country in question, with a specific form requesting the products. The prescription and accompanying documents may be required by law. Diagnoses, for which IVIG is approved, along with the associated international disease codes, are listed in Table 7.
Primary immunodeficiencies for which IVIG is indicated and their International Classification of Diseases codes (ICD).
| Disease: antibody deficiency with normal immunoglobulins | ICD: D80.6 |
| Disease: adenosine-deaminase deficiency | ICD: D81.3 |
| Disease: purine-nucleosideophosphorilase deficiency | ICD: D81.5 |
| Disease: class I MHC deficiency | ICD: D81.6 |
| Disease: class II MHC deficiency | ICD: D81.7 |
| Disease: IgG subclasses deficiency | ICD: D80.3 |
| Disease: hereditary hypogammaglobulinaemia | ICD: D80.0 |
| Disease: non-hereditary hypogammaglobulinaemia | ICD: D80.1 |
| Disease: transient hypogammaglobulinaemia of infancy | ICD: D80.7 |
| Disease: CVID with abnormal number of functional B cells | ICD: D83.0 |
| Disease: combined immunodeficiency with reticular dysgenesia | ICD: D81.0 |
| Disease: combined immunodeficiency with low number of B cells | ICD: D81.2 |
| Disease: combined immunodeficiency with low number of B and T cells | ICD: D81.1 |
| Disease: CVID with autoantibodies and B and T cells | ICD: D83.2 |
| Disease: immunodeficiency with high levels of IgM | ICD: D80.5 |
| Disease: other combined immunodeficiencies | ICD: D81.8 |
| Disease: other combined immunodeficiencies with defects in antibody production | ICD: D80.8 |
| Disease: other common variable immunodeficiencies | ICD: D83.8 |
| Disease: DiGeorge syndrome | ICD: D82.1 |
| Disease: Wiskott–Aldrich syndrome | ICD: D82.0 |
| Disease: Ataxia-telangiectasia | ICD: G11.3 |
In Latin America, the process of obtaining IVIG for treatment of PIDD varies from one country to another:
- •
In Brazil, the patient is responsible for obtaining IVIG and bringing it to the location where the infusion will be administered (http://www.mp.rs.gov.br/areas/dirhum/arquivos/medicamentosespecializados.pdf). Such locations may include: (1) hospitals, with admission for medical supervision; (2) day hospitals, with admission for medical supervision; or (3) medical clinics that have authorisation from the Agência Nacional de Vigilancia Sanitária (ANVISA) to perform infusions, with medical supervision. Availability of an appropriate setting to administer infusion is problematic in some regions of Brazil. The best option is a day hospital, but if this alternative is not available, a blood centre or oncology service may be used. The unit responsible for the infusion must maintain the medical record, check vital signs before and after the infusion, evaluate the need for premedication, and also record AEs as well as the product's name and batch number. The setting must be equipped and prepared to manage any infusion-related AEs. For patients with private insurance, obtaining IVIG is simple and it is generally administered in day hospital centres, instead of at home.
- •
In Mexico, IVIG is part of the “cuadro básico de medicamentos” from the Ministry of Health and the health system is responsible for having IVIG available to treat PIDD patients. Nonetheless, IVIG products are not always available, or they may be available in limited quantities that permit less than the prescribed doses needed for PIDD patients. In Mexico, most IVIG infusions are carried out in a hospital or medical clinic, with the patient admitted and under medical supervision.
- •
In Chile, IVIG treatment is not guaranteed by a health programme for PIDD. It is usually given by the hospital at which the patient is diagnosed. In general, IVIG is given to the patient at the time of diagnosis. However, in the public health system, further treatment with IVIG is not always continuous; it is sometimes discontinued for several weeks before it is administered again. In both the public and private systems, IVIG infusions are almost always administered on an inpatient basis with medical supervision in a setting equipped and prepared to manage any infusion-related AEs. In general, the immunologist determines whether or not the patient requires premedication.
- •
In Argentina, there have been no problems with IVIG product availability for paediatric patients since 2001; the government provides products to all patients treated at children's hospitals. Obtaining products for adults is sometimes more difficult, but these barriers can almost always be overcome. Legislation passed in 2009 and 2011 states that IVIG and other needed treatments should be available to patients with PIDD. In public hospitals of Buenos Aires, children with PIDD receive IVIG at no cost but other patients may be supplied by a social security system.
- •
In Peru, IVIG is paid for by the social security system. Two hospitals manage patients with PIDD—the Edgardo Rebagliati Martins National Hospital and Guillermo Almenara Irigoyen National Hospital; both are part of the social security system. Only 17 patients with PIDD were being treated in Peru in 2007, so these conditions are certainly underdiagnosed in this country of approximately 25 million people.
- •
In Costa Rica, patients diagnosed with PIDD with an indication for IVIG are treated at hospitals in the social security system; this therapy is included as part of services paid for by the health system. When patients reach adulthood, treatment is continued in adult hospitals. The product is administered at day hospitals on an outpatient basis.
- •
In Colombia, every citizen is entitled to a basic health plan subsidised by the government (POS, for Plan Obligatorio de Salud). This plan stratifies the population into two groups: low-income people are covered under a subsidised regimen and others are covered through a contributive regimen. The POS provides basic coverage for the diagnosis and treatment of PIDD93,94 for people in the subsidised regimen; however, users in the contributive regimen must purchase additional coverage through private healthcare providers. In addition, the POS has a component named POS Medicines that is defined by the Comisión de Regulación en Salud (CRES) and provides an annual list of essential medicines covered by health insurance providers. This list does not include haemoderivates such as IVIG or SCIG. Patients in need of immunoglobulin replacement therapy obtain treatment from their respective healthcare providers at no cost, but the prescribing physician must fill in a special form to be reviewed by a committee of experts that decides if the IVIG/SCIG is appropriate. Finally, gammaglobulin costs are refunded through the Fondo de Solidaridad y Garantia, FOSYGA, (the government's national fund) to the health insurance provider.
Infusion at home has become the treatment of choice for many patients receiving IVIG. Available results indicate that the numbers of infections, use of antibiotics, AEs, and levels of immunoglobulin achieved are all similar in patients infused at home or in the hospital.95–97 However, home administration of IVIG is not available in Latin American countries.
SCIGIn recent years, there has been great interest in the use of SCIG. This route of administration does not require venous access, which may be problematic in children and some adult patients. SCIG also reduces some of the complications that may occur with intravenous infusions.98,99 Administration of SCIG is performed at weekly intervals, which promotes better control of IgG levels versus monthly infusions of IVIG.99–101 Self-administration of SCIG at home without the requirement for a central line increases patient autonomy and well-being.102 Procedures for self-infusion of SCIG are learnt easily by both adults and children. This approach to delivering immunoglobulin replacement therapy is safe and has a low risk for systemic AEs.99,103
Adverse events noted most often with SCIG include oedema and erythema at the site of infusion, but these resolve within 12h after administration in most patients. Application of a warm compress may accelerate resolution of these events. Few patients require premedication for SCIG, and results to date indicate no risk for long-term tissue damage, fibrosis, or lipodystrophy at the site of infusion.104
The solutions prepared for SC infusion have IgG concentrations of 10%, 12%, 16%, or 20%.104 The recommended rate of infusion is 10mL/h, slowly increasing to 22mL/h if the patient does not experience AEs. Administration of SCIG often requires the use of an infusion pump.100 The product is loaded into a 10- or 20-mL syringe using a 1.9cm butterfly needle; the insertion is made at 90° or 45° into the abdominal skin.104 Infusions are delivered weekly, so if the patient receives 400mg/kg of IVIG every 28 days, he or she should be given SCIG at a dose of 100mg/kg/week. A volume of >20mL should not be administered at a single site; multiple sites must be used when higher volumes are required.97 The cost for SCIG administered at home is lower than that for IVIG delivered in the hospital, thus lowering the cost burden for the healthcare system.105–107
Several studies have demonstrated that regular infusion of SCIG is as effective as IVIG for the prevention of infection in children or adults with PIDD.108 SCIG is safe, increases a patient's well-being, provides more stable immunoglobulin levels, and has a lower cost compared with IVIG. Although physicians and patients should consider this choice of administration route,109 it is not available in all countries. For example, patients do not have access to SCIG in Chile and Brazil. At this time, Argentina has around 35 patients undergoing this form of treatment. The trend to infuse SCIG by push, rather than with infusion pumps, is especially welcome in Latin America and facilitates drug administration.
ConclusionsThe benefits of IVIG and SCIG for the prevention of infections in patients with antibody deficiencies who require replacement of antibodies are well established. Safe administration of these highly effective treatments requires understanding of product characteristics, common AEs, and patient-related factors that may influence product selection and the risks of side effects. Manufacturers’ instructions for infusion must be followed carefully and the interval between infusions established after the patient's third administration of IVIG. Non-serious AEs are common in patients receiving IVIG, and both their frequency and severity can be reduced by appropriate premedication. Potentially fatal reactions are rare and may be prevented by clinical supervision, early intervention, the interruption of the infusion when necessary, and provision of appropriate supportive care.
The authors wish to acknowledge Baxter for unrestricted educational grant support and BSG Communications for providing editorial assistance.
