


In this study, we aimed to discover potential gene targets for treating childhood asthmatics.
MethodsWith the microarray data downloaded from Gene Expression Omnibus (GEO) database, we explored the common differentially expressed genes (DEGs) in children with severe asthma and mild asthma (SA vs. MA) or healthy controls (SA vs. HC). Then we performed hierarchical clustering, function and pathway enrichment analysis for the common DEGs.
ResultsA total of 81 genes were identified to be differentially expressed in SA vs. MA and SA vs. HC group. Hierarchical clustering of the 81 DEGs could crudely separate the SA, MA and healthy individuals. The overrepresented GO terms of the common DEGs were related with lipid biosynthetic process (21.74%), pigment biosynthetic process (13.04%) and nucleoside monophosphate metabolic process (13.04%). Only one pathway was significantly enriched, which was the antigen processing and presentation pathway involved with CD4 and RFX gene.
ConclusionsThe antigen processing and presentation pathway and lipid biosynthetic process may play roles in the pathogenesis of severe asthma. CD4 and RFX provide a therapeutic possibility for childhood asthma.
Asthma is the most common chronic inflammatory disease in airways of children, and which has a significant effect on substantial daily lives of individuals.1 Asthma can result in variable and recurring symptoms including airflow obstruction, bronchospasm, chest tightness and shortness of breath.2,3 The combination of genetic and environment factors has been documented to contribute to the onset of asthma.4 The increasing trend in the prevalence of childhood asthma has been reported since 1980s.5 It is reported that approximate 300 million people suffered asthma worldwide in 20116 and there are around 250,000–345,000 deaths attributable to this disease.7 For the high mortality, hospitalization rates and health care costs, asthma has become a health problem all over the world.8,9
Currently, the main therapy for asthma is the use of daily, long-term controller and quick-relief medications. The quick-relief medications are primarily applied for acute symptom treatment and long-term control medicines are used to prevent exacerbation. Despite the potent positive effects of drugs are available for asthma patients, high dose or long-term use of medicine may remain adverse effects such as development of cataracts, impaired growth in children and decrease in bone mineral density.10,11 Numerous studies have been conducted to investigate the mechanism underlying asthma progression and explore the potential alternative therapies for asthma in children. A previous study shows that reactive oxygen species (ROS) generated in airway cells play key roles in enhancing the inflammation in the development of asthma.12 Oxidative stress and gene transcription are targets in antioxidant therapy for asthma. In addition, some cytokines and proteins are determined to be therapeutic targets for asthma, such as Th2 cytokines, nterleukin-9, chemokine receptors, histone deacetylase.13–15 However, studies concerning the gene targets for asthma seem to be insufficient.
In the present study, we explored the differentially expressed genes (DEGs) in children with severe asthma and mild asthma and healthy controls. The significant functions and pathways associated with DEGs were further investigated. The purpose of our study was to screen the potential gene targets for asthma treatment and provide new insight to the effective management of childhood asthma.
Materials and methodsAffymetrix microarray dataThe microarray data of GSE27011 was downloaded from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). The gene expression patterns analyzed in this work were developed from 54 white blood cell samples, including 17 samples from children with severe therapy-resistant asthma, 19 from mild asthmatic children and 17 from healthy individuals. Patients with severe and mild asthma were confirmed by extensive clinical and immunological characterization. The array data was originally developed by Orsmark-Pietras et al.16. We downloaded the raw data and the annotation files for further analysis based on the platform of Affymetrix Human Gene 1.0 ST Array.
Data process and DEGs analysisBased on the annotation files, a total of 33297 probes were corresponded to gene symbols. After the probe level data were converted to gene expression values, all the expression data were normalized using robust multiarray average (RMA)17 algorithm. Differentially expressed genes (DEGs) between children with severe asthma and mild asthma (SA vs. MA) or healthy control (SA vs. HC) were analyzed by limma package in R programming language.18 The Benjamini-Hochberg method was used to adjust for the false discovery rate.19 DEGs were displayed with false discovery rate (FDR) and FDR<0.05 was defined as the cut-off value. In addition, we used an interactive visualization method20 to present the DEGs in both SA vs. MA and SA vs. HC group and those specific in SA vs. MA or SA vs. HC group.
Hierarchical clustering of DEGsHierarchical clustering has been widely applied in analysing gene expression patterns.21,22 With hierarchical clustering, the most similar expression patterns can be clustered in a hierarchy of nested subsets. The expression-level data of mutual DEGs in both SA vs. MA and SA vs. HC group were collected based on the raw files. The hierarchical clustering analysis23,24 of the expression data was performed by pheatmap package in R (http://cran.r-project.org/web/packages/pheatmap/index.html) based on Euclidean distance.25 Hierarchical clustering analysis of the expression data of common DEGs showed advantage in determining whether the screening DEGs had sample specificity or not. Results of hierarchical clustering were displayed by using heat map.
Function and pathway enrichment analysis of DEGsThe Database for Annotation, Visualization and Integrated Discovery (DAVID) is a gene functional classification tool that provides a comprehensive set of functional annotation tools for investigators to understand biological meaning behind large list of genes.26 The Kyoto Encyclopedia of Genes and Genomes (KEGG) database is a collection of biochemical pathways, which connects the higher-level complexity of cellular processes and organism behavior with a large scale of genes.27,28
The Gene Ontology (GO) annotation and KEGG pathway enrichment analysis for mutual DEGs in SA vs. MA and SA vs. HC group were performed using the functions of DAVID.29,30P<0.05 was set as the cut-off value. The percentage of significantly enriched GO terms of DEG sets were visualized in three dimensional pie charts with the application of plotrix package in R.31
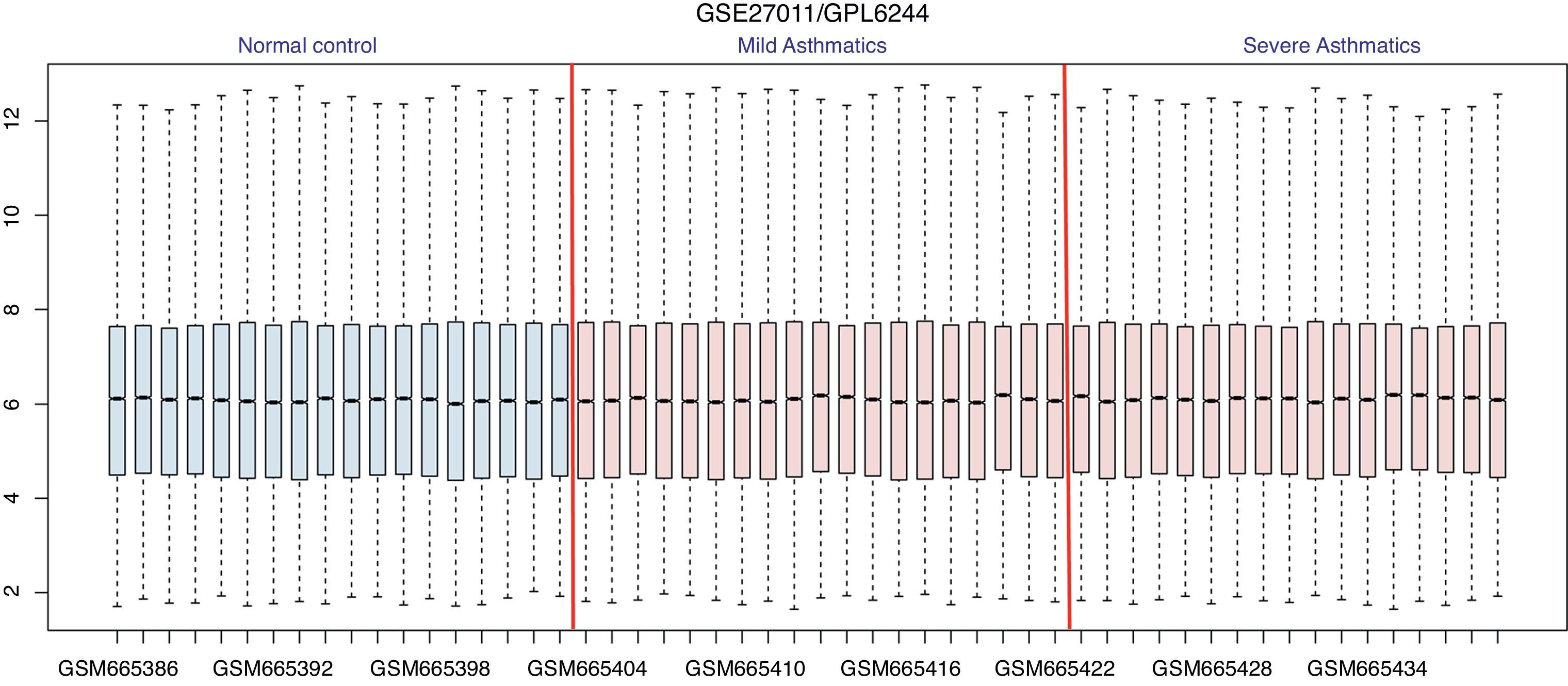
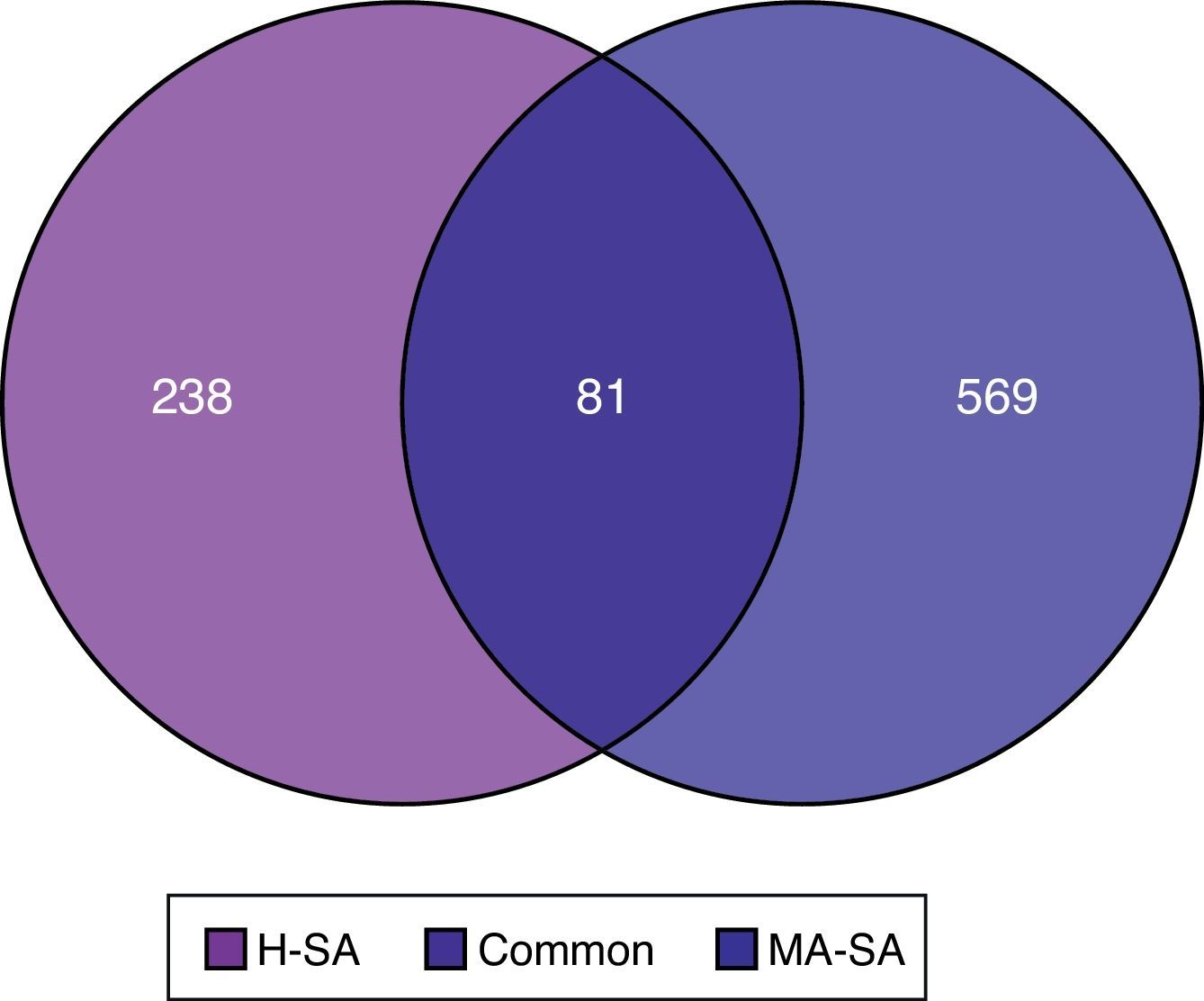
ResultsIdentification of DEGsAfter pre-processing, the obscuring variations in raw expression data were normalized (Fig. 1). Then we performed DEGs analysis by limma package. Using the threshold value of FDR<0.05, a total of 969 genes were determined to be differentially expressed, among which 319 DEGs were validated in SA vs. HC group and 650 in SA vs. MA group. As shown in Fig. 2, there are 81 common DEGs in SA vs. HC and SA vs. MA group. In addition, total 569 genes are specific DEGs in SA vs. MA group and 238 DEGs are only in SA vs. HC group.

Box plots of data normalization. The x-coordinate represents samples; y-coordinate represents gene expression values. The blue box plots are samples from healthy controls and the red ones are samples from patients with asthma. The black line in the box plots represent expression median.

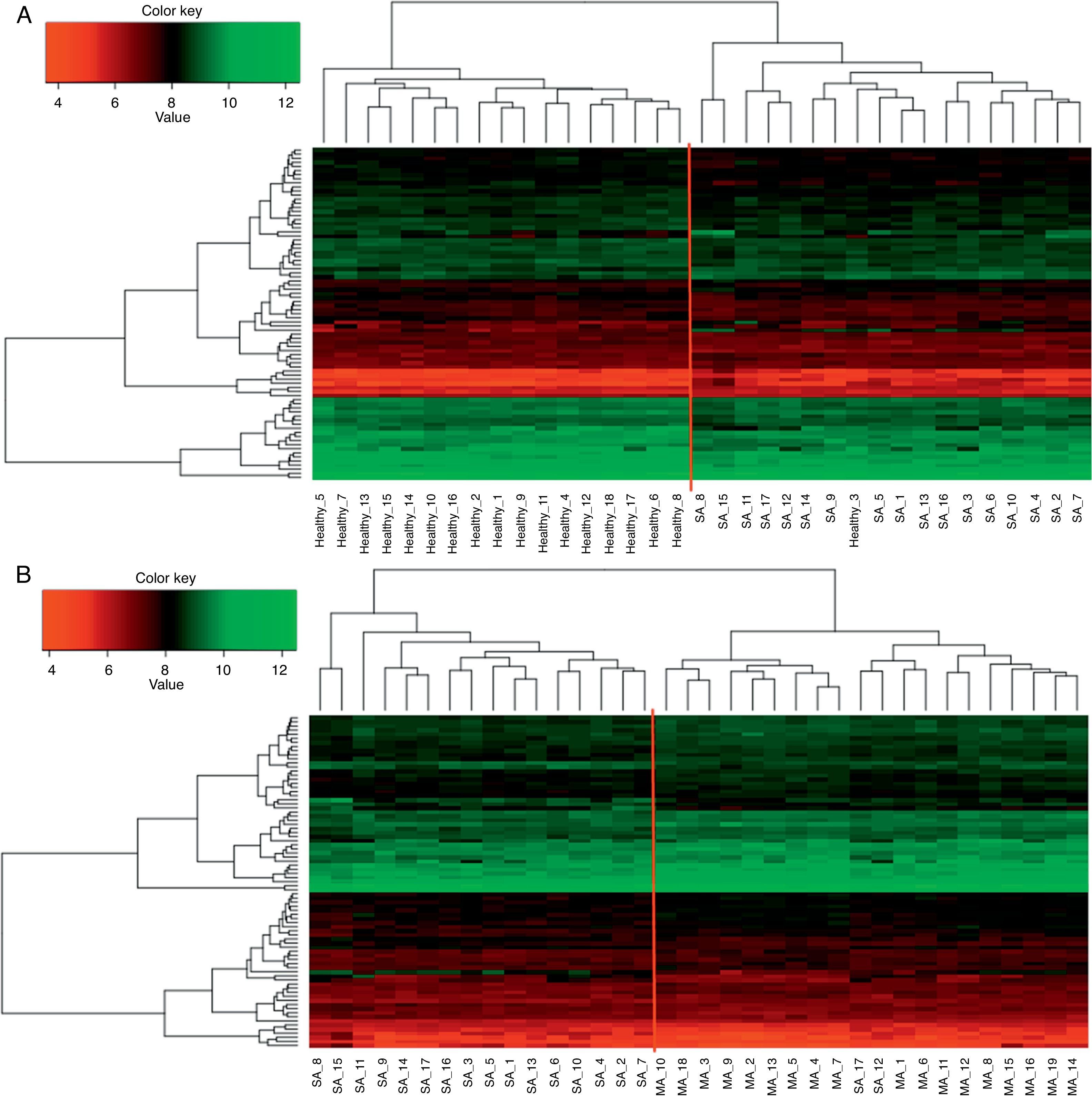
In order to present the orderly choreography of expression program, we performed the hierarchical clustering for the expression data of common DEGs. As shown in Fig. 3, samples from SA, HC and MA group were relatively clearly distinguished based on the expression data of common DEGs, suggesting that the DEGs identified in our paper were significant.
Function annotation and pathway enrichment analysis of common DEGs
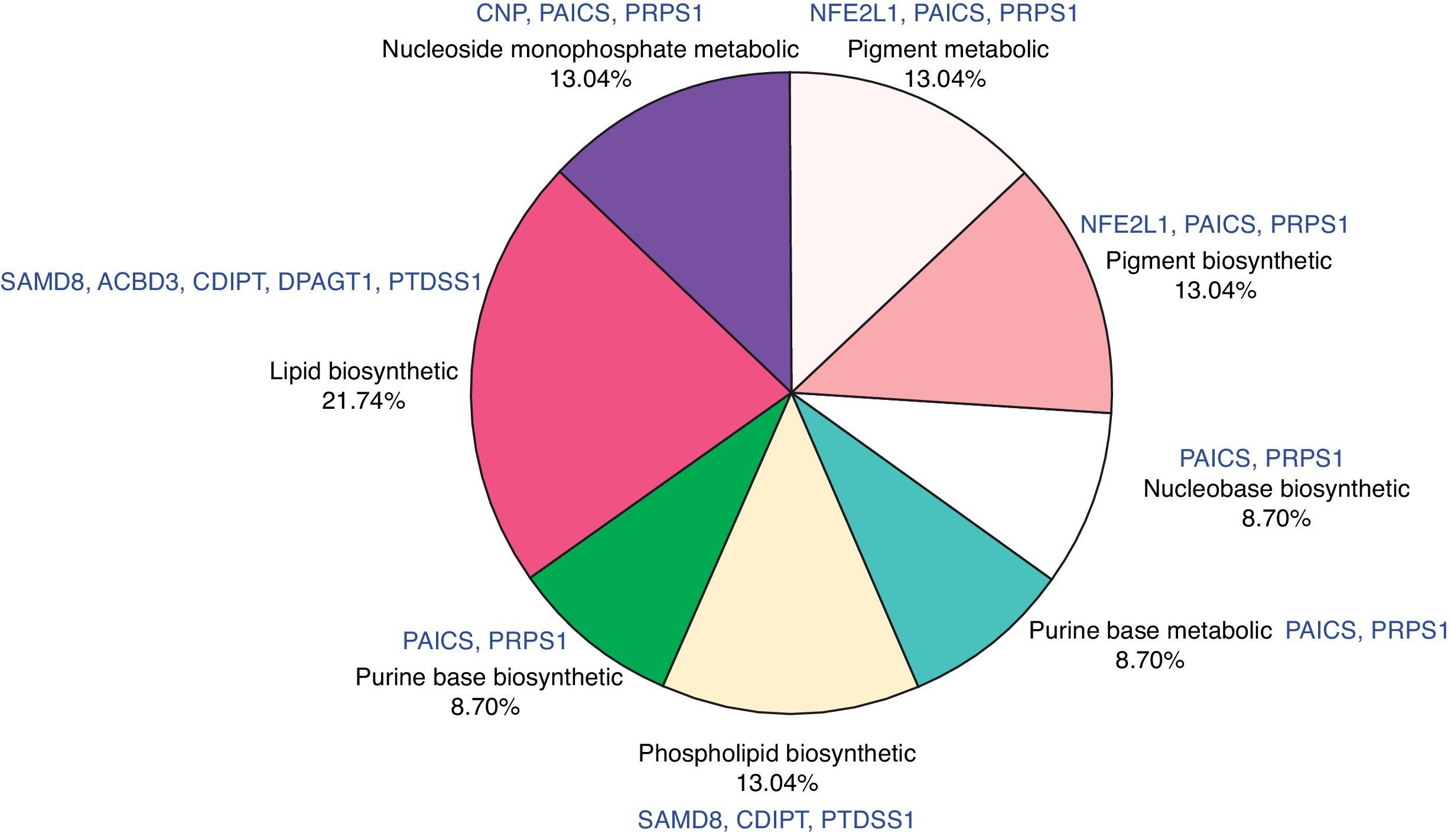
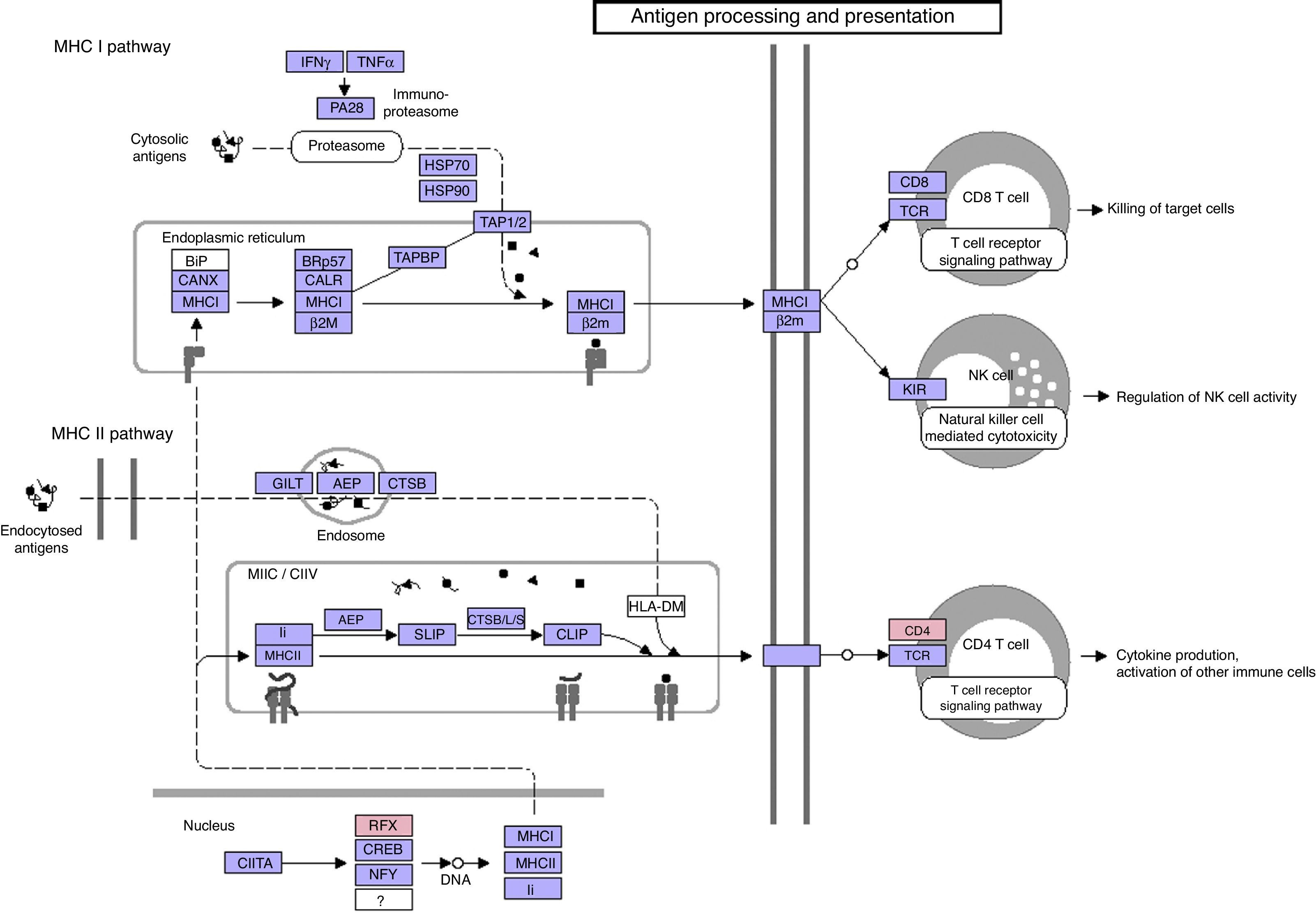
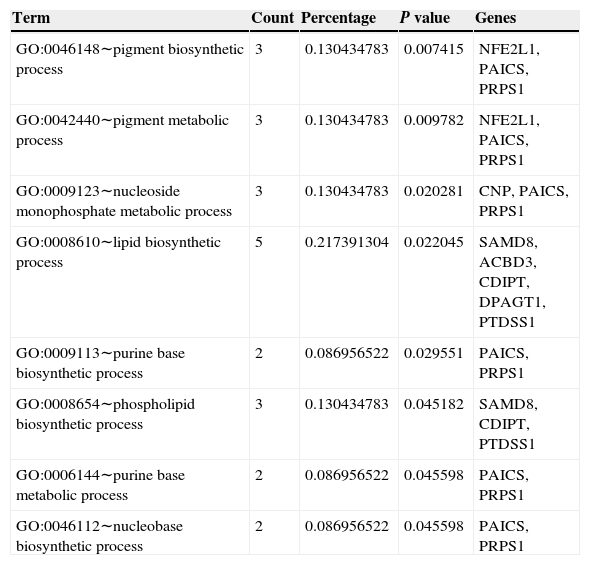
In order to investigate the common DEGs in functional level, we performed GO and KEGG enrichment analysis. Total 8 GO terms were significantly enriched by the common DEGs, such as pigment biosynthetic process, nucleoside monophosphate metabolic process and lipid biosynthetic process (Table 1). The percentages of overrepresented GO terms of DEGs were shown in Fig. 4. The lipid biosynthetic process accounted for the largest proportion (21.74%). Pigment biosynthetic process, pigment metabolic process, nucleoside monophosphate metabolic process and phospholipid biosynthetic process represented a proportion of 13.04%. Only one pathway was significantly enriched by DEGs such as CD4 and RFX, which was named as antigen processing and presentation (P=0.0038) (Fig. 5).
Results of function enrichment analysis of differentially expression genes.
| Term | Count | Percentage | P value | Genes |
|---|---|---|---|---|
| GO:0046148∼pigment biosynthetic process | 3 | 0.130434783 | 0.007415 | NFE2L1, PAICS, PRPS1 |
| GO:0042440∼pigment metabolic process | 3 | 0.130434783 | 0.009782 | NFE2L1, PAICS, PRPS1 |
| GO:0009123∼nucleoside monophosphate metabolic process | 3 | 0.130434783 | 0.020281 | CNP, PAICS, PRPS1 |
| GO:0008610∼lipid biosynthetic process | 5 | 0.217391304 | 0.022045 | SAMD8, ACBD3, CDIPT, DPAGT1, PTDSS1 |
| GO:0009113∼purine base biosynthetic process | 2 | 0.086956522 | 0.029551 | PAICS, PRPS1 |
| GO:0008654∼phospholipid biosynthetic process | 3 | 0.130434783 | 0.045182 | SAMD8, CDIPT, PTDSS1 |
| GO:0006144∼purine base metabolic process | 2 | 0.086956522 | 0.045598 | PAICS, PRPS1 |
| GO:0046112∼nucleobase biosynthetic process | 2 | 0.086956522 | 0.045598 | PAICS, PRPS1 |


The increasing prevalence of asthma affects public heath, especially the overall quality of life in children. Due to improper measures and delayed treatment time, a large cohort of asthmatic children developed to severe uncontrolled asthma.32 Children with asthma are at high risk for influenza-related complications. The effective management guidelines for childhood asthmatics have been highlighted worldwide. In this work, we analyzed the microarray data of white blood samples from children with severe asthma, mild asthma and healthy controls. In this paper, we aimed to explore the mechanism of asthma progression and discovery the potential gene targets for childhood asthma treatment. Results showed that a total of 81 genes were differentially expressed in severe asthmatics compared with children with mild asthma and controls. The hierarchical cluster analysis of the 81 DEGs indicated that the DEGs were specific in childhood severe asthmatics that differed from those in children with SA and MA. To explore the potential biological function for 81 DEGs, we performed GO and pathway enrichment analysis.
Our results revealed that antigen processing and presentation pathway (P=0.0038) were significantly enriched by 81 DEGs. It is well documented that environment and gene factors are the main causes for the development of asthma.33 In early childhood, it is common for children to present the initial sensitization to airborne environmental allergens.34 Harmless inhaled antigens (allergens) are responsible for an asthmatic reaction that may progress to persistent atopic asthma for several years. The inhalation of allergens leads to the sensitization of T helper (Th) type 2 cells induced by dendritic cells (DCs).35 A subtype of DCs took up antigens and played a critical role in antigen processing and presentation, which further stimulated T cells and inflammation response.36 For allergen-induced airway hyperreactivity and chronic inflammation to be the primary characteristics in asthma development, antigen processing and presentation may be an critical event in the progression of persistent inflammation in the airway wall that leads to structural and functional changes in local tissues and symptom development.
In addition, our results revealed that CD4 and RFX were involved in antigen processing and presentation pathway (Fig. 5). CD4 encoding CD4 molecules have been found to be associated with inflammation and cell immune response.37 CD4 molecules are mainly expressed on helper T cells and have functions in helping T cell receptor to identify antigen and promoting signal transduction of T cell activation.38,39 Therapy targeting CD4 expression may be potent in inhibiting antigen presentation and suppressing chronic inflammatory response. Regulatory factor X (RFX) transcription factors have been determined to be involved in the development of a large list of serious human diseases.40 Although evidence concerning the key role of RFX in the development and progression of childhood asthma is rare, further studies of RFX may hold promise for gaining a novel insight to effective treatment of childhood asthma.
Furthermore, the biological process of lipid biosynthetic process was found to be overrepresented by DEGs with the largest percentage (21.74%). As outlined in previous studies, lipid peroxidation was determined to be related with the severity of asthma in children.41 Lipid peroxidation elicited by the increasing generation of reactive oxygen species in asthma contributed to the dysfunction of airway.42 The lipid metabolism was revealed to be changed in children with asthma after treatment by budesonide. Besides, lipid extract of New Zealand green-lipped mussel was found to be effective in treating asthma by inhibiting eicosanoids production.43 All these implied that lipid played a crucial role in the pathogenesis of asthma but the lipid biosynthetic process needed to be further investigated in asthma progression.
In summary, DEGs specific in childhood severe asthmatics were closely associated with the development of severe asthma. The antigen processing and presentation pathway and lipid biosynthetic process played a role in the pathogenesis of severe asthma. CD4 and RFX may be potential therapeutic targets for asthma. Our paper provided new insights to discover the effective therapy for childhood asthma. However, a large number of studies should be conducted to confirm our results with sufficient experimental evidence.
Conflict of interestAll authors declare that they have no conflict of interests to state.
Ethical disclosuresProtection of human subjects and animalsThe authors declare that no experiments were performed on humans or animals for this investigation.
Patients’ data protectionConfidentiality of Data. The authors declare that no patient data appears in this article.
Right to privacy and informed consentThe authors declare that no patient data appears in this article.
