




Myotonic dystrophy type 1, also known as Steinert's disease, is a multisystemic disorder with significant genetic and clinical heterogeneity. Apart from skeletal muscles' myotonia and wasting, a variety of system organs can be affected. We report on a 49 years old female patient with unremarkable medical and family history, who presented with elevated liver enzymes without signs or symptoms of chronic liver disease neither neurological features. Initial assessment, including liver biopsy, did not reveal the cause of these abnormalities. Eight months later, she complained for disequilibrium and eventually electromyography confirmed the diagnosis of Steinert's disease. Steinert's disease should be considered in the differential diagnosis of patients with elevated liver enzymes, as long as abnormal liver tests may be the initial presentation. The pathophysiological mechanism of this abnormality remains unclear.
Steinert’s disease (myotonic dystrophy type 1; MD) is an autosomal dominant multisystemic disorder with remarkable genetic heterogeneity, which is considered the most common form of adult-onset muscular dystrophy. Myotonic dystrophy affects skeletal muscles, particularly facial muscles, distal muscles of the forearm and ankle dorsiflexors leading to weakness, myotonia (slowed relaxation after muscle contraction) and muscle pain. Considering its multisystemic pattern, several other organ systems are involved such as heart, gonads, respiratory and gastrointestinal tract, endocrine system, central and peripheral nervous system. Consequently, patients may be presented with variable clinical features. As long as there is no disease-specific treatment available for Steinert’s disease, the management of these patients depends on clinical presentation and symptoms.
Abnormal results of liver function tests have been reported in this setting but the pathophysiological mechanism is still unknown. We report on a patient who presented with elevated liver enzymes without indications of chronic liver disease, months before the diagnosis of Steinert’s disease.
Case ReportA 49-year-old Caucasian female was seen at the outpatients-clinic because of abnormal liver function tests. Apart from debilitation, for a period of 3 months, she had been asymptomatic. She reported no alcohol consumption and she had given up smoking one month ago following a thirty years-smoking history. She was receiving thyrormone (T4) as hormone replacement therapy (partial thyroidectomy due to a thyroid nodule 4 years ago) and medication (risedronate, alfacalcidol) for osteoporosis. She had also been operated twice for uterine fibromyomas 16 and 7 years ago, whereas her family history was unremarkable.
Clinical examination revealed a slim, tall woman (BMI 18, 3 kg/cm2) without neurological findings or signs of chronic liver disease. Laboratory tests performed showed:
- •
Alanine aminotransferase (ALT) 163 U/L (normal range between 0-45).
- •
Aspartate aminotransferase (AST) 131 U/L (normal range 5-46).
- •
Gamma glutamyl-transferase (GGT) 314 U/L (normal range 5-32).
- •
Alkaline phosphatase (ALP) 414 U/L (normal range 100-290).
- •
Total bilirubin 0.29 mg/dL (normal range 0, 1-0, 3).
- •
Cholesterol 279 mg/dL (normal range 140-220).
Autoimmune screening was negative (AMA, ASMA, ANA, ANCA, anti-LKM) apart from a slight reduction of immunoglogulin M (IgM 36 mg/dL, normal range 40-300) and a great one of immunoglobulin G (IgG 339 mg/dL, normal range 700-1600).
Tests for viral hepatitis and metabolic screening (a1-antitrypsin, caeruloplasmin, ferritin) were negative. IgM antibodies for viral infections (EBV, HSV I and II, CMV), screening for HIV 1, 2, anti-gliandin and transglutaminase antibodies were also negative. Thyroid function tests were between normal ranges.
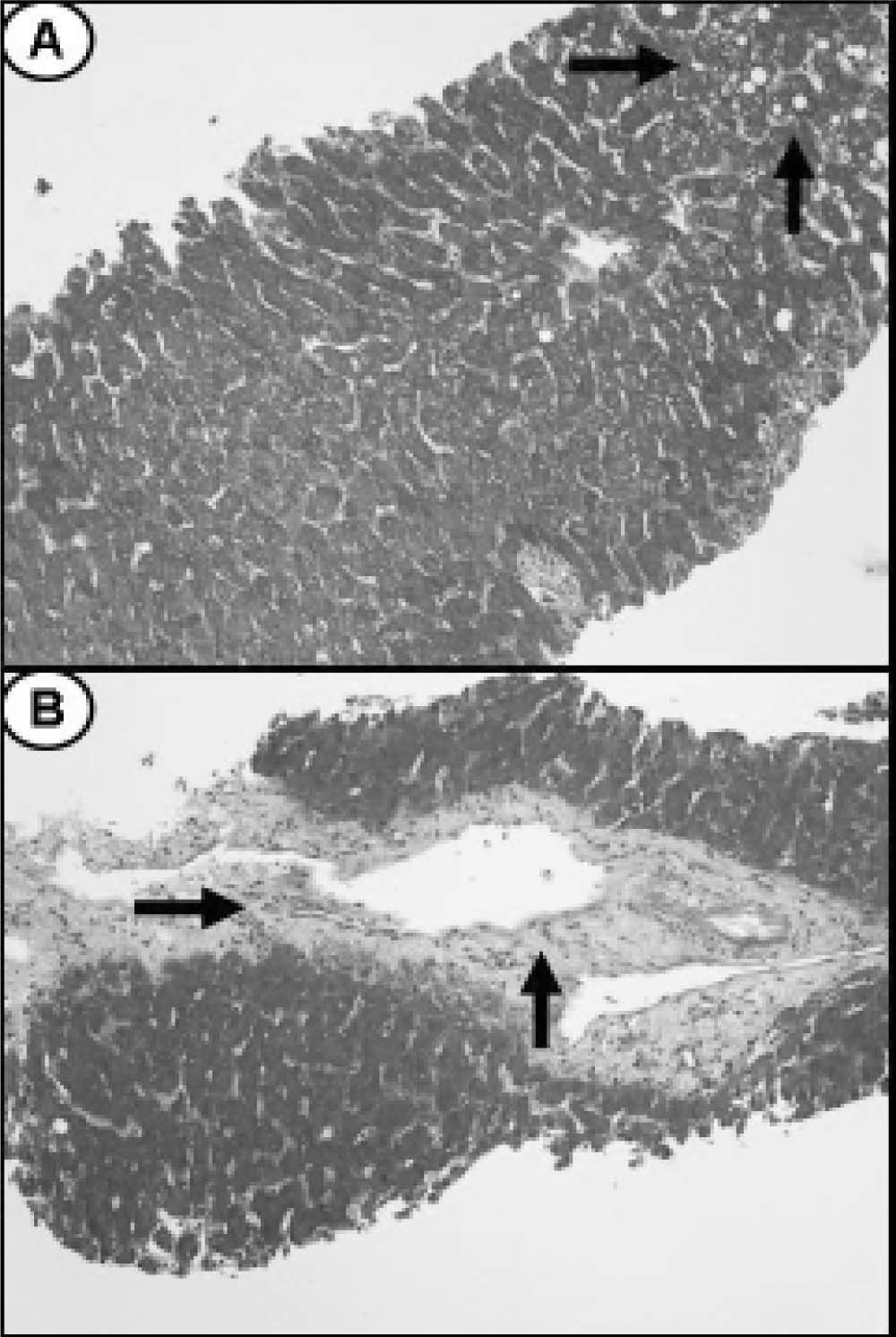
Ultrasound of upper abdomen showed no abnormal findings except from fatty liver infiltration. Magnetic retrograde cholangiopancreatography (MRCP) and computer tomography (CT) were also normal. A liver biopsy (Figure 1) showed 5% steatosis, pericentric fibrosis, fibrous dilation of portal spaces and focuses of pericellular and bridging fibrosis, with absence of necroinflammation in portal spaces and lobules, neither microscopic disorders of small bile ductules; findings unable to correlate with a particular disease. No medication was prescribed and she was followed up regularly.
 (H&E × 250). B. Microphotograph showing an area of bridging fibrosis in the liver biopsy (arrows) (H&E × 250).")
Eight months later she was presented with disequilibrium. The physical examination revealed slight ptosis of the eyelids, myotonia noted in face and jaw muscles, weakness of the neck muscles, distal upper limbs and lower limbs. Laboratory tests demonstrated:
- •
ALT 134 U/L.
- •
AST 102 U/L.
- •
GGT 142 U/L.
- •
ALP 107 U/L.
- •
Total bilirubin 0.29 mg/dL.
- •
Creatinine kinase was 295 U/L (normal range 0 to 142 U/L).
Tests for multiple myeloma (radiography for bone lytic lessions, urinary light chain excretion, beta-2 microglobulin, protein electrophoresis) were negative whereas the levels of vitamin 12, folic acid and aldolase were within normal range. Electromyography confirmed the diagnosis of myotonic dystrophy type 1 or Steinert disease. After diagnosis, she went through a cardiac echocardiography which did not show any heart conduction or structural abnormality. Spirometry showed the presence of a mild restrictive abnormality. Sixteen months later from her initial visit, patient remains in good clinical condition, without worsening of her neurological condition, whereas liver enzymes persist to be elevated (AST 58 U/L, ALT 48 U/L, GGT 193 U/L and ALP 329 U/L).
DiscussionIn the literature there are reports 1-5 that indicate liver involvement in already diagnosed MD (table 1). We present a case of Steinert’s disease with abnormal liver function tests months before the presentation of clinical symptoms. Consequently physicians should include MD-although rarein the differential diagnosis of elevated liver and cholostatic enzymes. The pathophysiology should be further evaluated.
Liver involvement in myotonic dystrophy type 1.
| Author | Year of publication | Type of publication | Main findings |
|---|---|---|---|
| Ronnemaa T, et al.1 | 1987 | Prospective study | Elevated GGT in 65% patients with MD. |
| Achiron A, et al.2 | 1998 | Prospective study | Abnormal liver enzymes in 66% patients with MD. |
| Heatwole C, et al.3 | 2006 | Prospective study | Abnormal values in all liver enzyme tested; 45% elevated GGT. |
| Siciliano G, et al.4 | 2005 | Prospective study | Elevated GGT and advanced oxidation protein products (AOPPs) in patients with MD1; possible role of oxidative stress in the pathogenesis. |
| Syn JK, et al5 | 2009 | Case presentation | Elevated liver enzymes years before the onset of Steinert’s disease in a 53 years old male. |
Myotonic dystrophy (MD) is a multisystemic, autosomal disorder with genetic and clinical heterogeneity. There are two forms, DM1 also called Steinert’s disease and DM2 which is generally milder in presentation with a later onset. DM1 is caused by an expansion of CTG trinucleotide repeat in the 3’-untranslated region of the dystrophia myotonica protein kinase gene (DMPK) on chromosome 19q 13.3,6-10 whereas DM2 results from an expansion of CCTG tetranucleotide repeat expansion located in intron 1 of the zinc finger protein 9 gene (ZNF9) on chromosome 3q 21.3.11 In each case, the genes are transcribed to RNA but are not translated.12 The mutant RNA results in abnormal function of several genes13 such as the skeletal muscle choride channel,14 the insulin receptor15 and cardiac troponin T.16 Both forms are presented as muscle weakness, myotonia, cataract, cardiac conduction abnormalities, insulin resistance, primary hypogonadism, hypogammaglobulinemia and respiratory abnormalities. Gastrointestinal manifestations include colicky abdominal pain, constipation, diarrhea, pseudo-obstruction and swallowing difficulties, all related to smooth muscle involvement.17
There is some evidence of liver involvement in MD but the pathophysiology is still uncertain. Ronnemaa, et al.1 studied the activity of serum GGT in 17 patients with MD. GGT activity was found elevated in 11 (65%) patients whereas most of them had one or more other pathological liver tests. In a later study,2 the authors evaluated prospectively liver and gallbladder function in 53 patients with MD. Abnormal activity of at least one liver enzyme was found in 35 patients (66%). Liver abnormalities were not correlated to disease severity or CTG expansion. Heatwole, et al.3 studied 126 MD patients mild to moderately affected. Patients demonstrated abnormal values in all liver enzyme levels tested (lactate dehydrogenase-LDH, GGT, AST, ALT and ALP).
It has been suggested that abnormal liver tests represent a cell membrane defect affecting the contractility of bile canaliculli and bile ductules.18,19 An alternative explanation is that the elevation of GGT results from dysfunction of the hepatocytes whereas the elevation of ALT, AST and LDH results from abnormal skeletal muscle.3 In another study,4 authors suggested that oxidative stress may play a role in the pathogenesis of MD. They investigated 39 patients with MD to assess the plasma concentration of advanced oxidation protein products (AOPPs) and GGT and related them to clinical severity scores. GGT and AOPPs were significantly higher in patients than in controls and, moreover, a correlation was found between serum GGT levels and AOPPs as well as between AOPPs and extra-muscular signs of the disease. GGT is known for catalyzing free radical formation and oxidative damage20 and its increase could result from the oxidative stress status.
To conclude, our patient presented with elevated liver enzymes without indications of chronic liver disease, months before the diagnosis of Steinert’s disease. Consequently, we demonstrate that abnormal liver tests may be the presenting feature of MD and this disorder should be included in the differential diagnosis of elevated liver enzymes. Moreover, considering these abnormal values as indications of liver dysfunction, medications undergoing metabolic clearance should be prescribed cautiously in this setting.



