



The primary bile acids (BAs) are synthetized from cholesterol in the liver, conjugated to glycine or taurine to increase their solubility, secreted into bile, concentrated in the gallbladder during fasting, and expelled in the intestine in response to dietary fat. BAs are also bio-transformed in the colon to the secondary BAs by the gut microbiota, reabsorbed in the ileum and colon back to the liver, and minimally lost in the feces. BAs in the intestine not only regulate the digestion and absorption of cholesterol, triglycerides, and fat-soluble vitamins, but also play a key role as signaling molecules in modulating epithelial cell proliferation, gene expression, and lipid and glucose metabolismby activating farnesoid X receptor (FXR) and G-protein-coupled bile acid receptor-1 (GPBAR-1, also known as TGR5) in the liver, intestine, muscle and brown adipose tissue. Recent studies have revealed the metabolic pathways of FXR and GPBAR-1 involved in the biosynthesis and enterohepatic circulation of BAs and their functions as signaling molecules on lipid and glucose metabolism.
Bile acids (BAs), the major lipid components of bile, are synthetized from cholesterol in the liver and subsequently conjugated to taurine or glycine, leading to an increase in their solubility. Immediately after synthetization, BAs are secreted into bile, as well as concentrated and stored in the gallbladder. Upon food intake, the gallbladder is stimulated by the entero-hormone cholecystokinin (CCK) to release bile into the duodenum, where BAs aid in the digestion and absorption of lipids and fat-soluble vitamins.1 Fasting serum BAs concentrations in healthy subjects are 0.2-0.7 μM to increase to 4-5 μM after each meal.2.5 Most of the BAs in the ileum are reabsorbed and return to the liver through the portal vein, and consequently, hepatic BA synthesis is inhibited by a negative feedback regulatory mechanism. However, BAs that escape from intestinal reabsorption enter the colon, where they are further transformed into the secondary and, more hydrophilic BAs, by the resident gut microbiota.6
The role of bile in lipid metabolism goes beyond that of fat emulsifier. Recent studies have also pointed to BAs as signaling molecules with metabolic effects via interaction with the nuclear receptors farnesoid X receptor (FXR), pregnane X receptor (PXR), and vitamin D receptor (VDR), G-protein coupled receptors such as GPBAR-1, and cell signaling pathways such as c-Jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK). Through these interactions, BAs help to regulate energy, glucose, lipids and lipoprotein metabolism.7
In this review, we summarize recent advances in the complex BAs physiology, focusing on novel findings about the regulatory mechanisms of BAs that are dependent and independent of nuclear FXR. We also briefly discuss the interaction between host microbiota and dietary intake on BA metabolism.
Biosynthetic Pathways of Bile Acids (Bas)BAs belong to a family of closely related acidic sterols synthesized from cholesterol, and represent the main catabolic pathway of cholesterol metabolism in humans. BAs are classified as soluble amphiphiles because of the ionized carboxylate or sulfonate group on the side chain that makes BAs water-soluble. In general, BAs possess a steroid nucleus of four fused hydrocarbon rings with polar hydroxyl functions. De novo BA biosynthesis occurs in the liver as the “primary” BAs, i.e., cholic acid (CA) and chenodeoxycholic acid (CDCA). Afterwards, the water solubility of BAs is increased by conjugation to either taurine or glycine, followed by hepatic secretion of BAs into bile and release by the gallbladder into the duodenum after the meal.8 The aliphatic side chain is conjugated in amide linkage (N-acyl amidation) with glycine or taurine at a ratio of 3:1 to increase water solubility of BAs (glycine > taurine) in bile and reduce BA toxicity (Figure 1).2 In bile, BAs act as cholesterol carriers together with phospholipids, and in the intestine, BAs act as surfactants and help the digestion and absorption of dietary cholesterol, triglycerides, and fat-soluble vitamins.9 Thus, BAs are essential in biliary cholesterol secretion and transport in bile, as well as hepatic catabolic products of endogenous cholesterol.
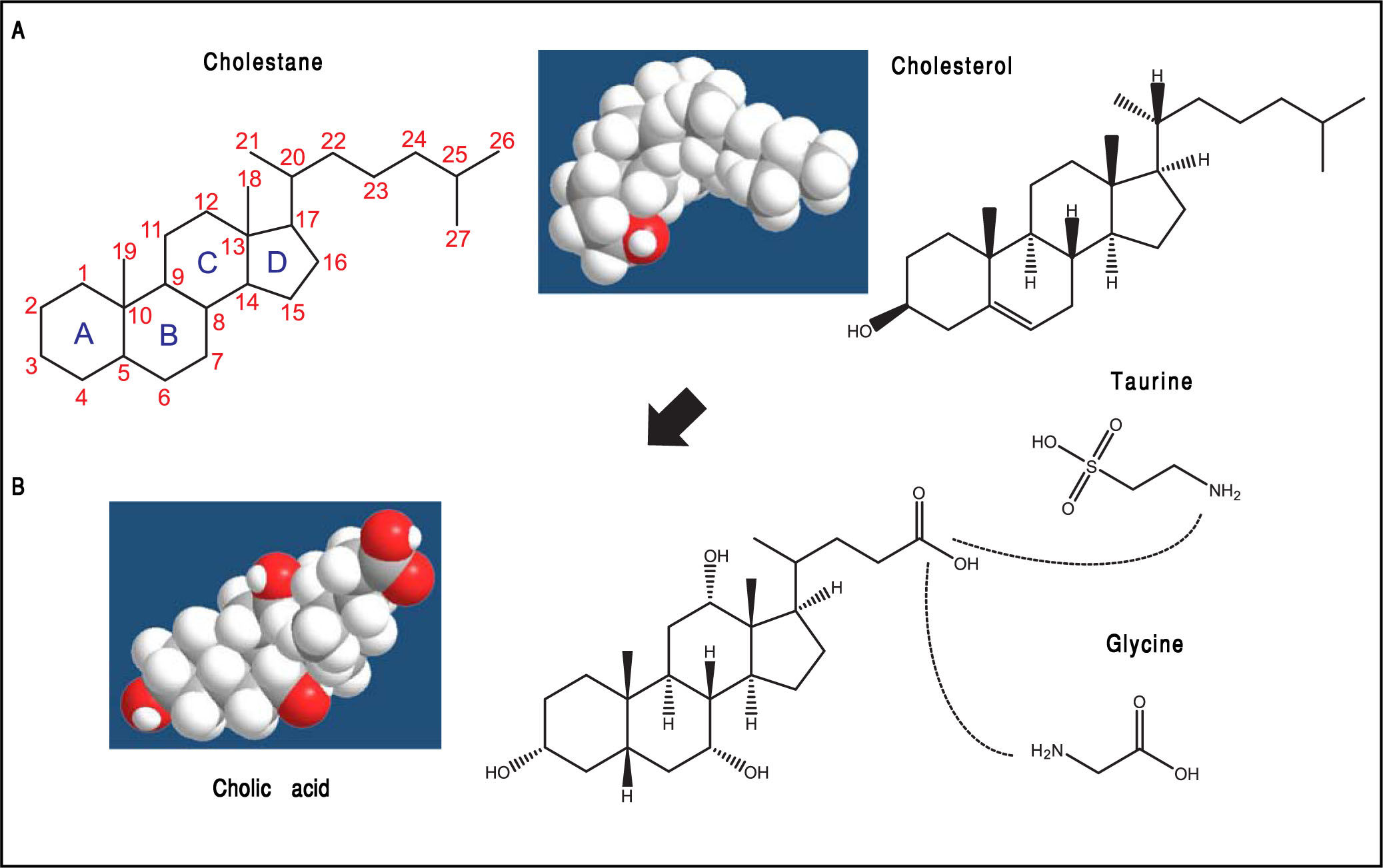
 is shown with numbering of the carbon atoms. The four fused hydrocarbon rings are labelled as A, B, C, and D. Cholesterol is shown as 3D structure and chemical formula. B. Hepatic cholesterol in the body is catabolized to bile acids (BAs), and cholic acid (CA) is shown as an example. CA possesses a steroid nucleus of four fused hydrocarbon rings with polar hydroxyl functions and an aliphatic side chain in amide linkage with taurine or glycine (dotted lines). The two enzymes involved in this process are the BA CoA synthase and the BA-CoA-amino acid N-acetyltransferase. The hydrophilic (i.e., polar) areas of BAs are the hydroxyl groups (-OH) (orientation of the hydroxyls: 3α, 7α, 12α) and conjugation side chain of either glycine or taurine. The hydrophobic (i.e., nonpolar) area is the ringed steroid nucleus.")
The general structure of cholestane (classified as a saturated 27-carbon tetracyclic triterpene) is shown with numbering of the carbon atoms. The four fused hydrocarbon rings are labelled as A, B, C, and D. Cholesterol is shown as 3D structure and chemical formula. B. Hepatic cholesterol in the body is catabolized to bile acids (BAs), and cholic acid (CA) is shown as an example. CA possesses a steroid nucleus of four fused hydrocarbon rings with polar hydroxyl functions and an aliphatic side chain in amide linkage with taurine or glycine (dotted lines). The two enzymes involved in this process are the BA CoA synthase and the BA-CoA-amino acid N-acetyltransferase. The hydrophilic (i.e., polar) areas of BAs are the hydroxyl groups (-OH) (orientation of the hydroxyls: 3α, 7α, 12α) and conjugation side chain of either glycine or taurine. The hydrophobic (i.e., nonpolar) area is the ringed steroid nucleus.
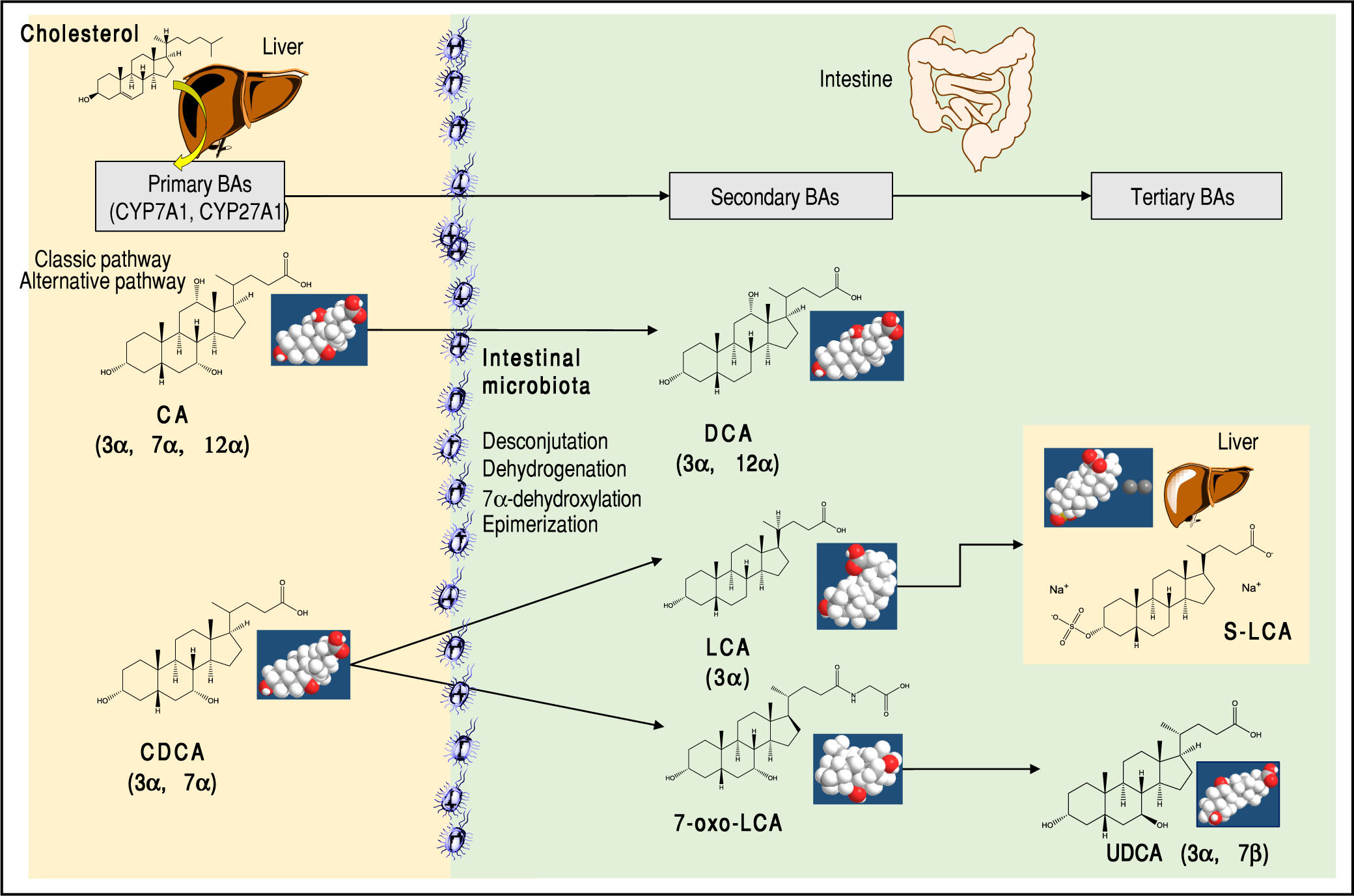
About 15% of conjugated BAs escape the absorption of the terminal ileum and enter the colon, where the resident gut microbiota promotes the deconjugation and bi-otransformation of the primary BAs into the secondary BAs such as deoxycholic acid (DCA) and lithocholic acid (LCA) and the tertiary BAs such as ursodeoxycholic acid (UDCA), as shown in Figure 2. Approximately 50% of DCA and a small amount of LCA and UDCA are re-ab-sorbed in the terminal ileum and colon and return to the liver via the portal vein. They enter the liver with assistance of sodium taurocholate cotransporting polypeptide (NTCP) transporter and organic anion transporting polypeptide (OATP) transporter. The secondary BAs are reconjugated with taurine or glycine. By contrast, all BAs in feces are deconjugated and consist mainly of DCA and LCA.
 and the dihydroxy chenodeoxycholic acid (CDCA). Two bio synthetic pathways are involved: the classical pathway is initiated by 7α-hydroxylase (CYP7A1) which stimulates the 7α-hydroxylation of cholesterol with synthesis of 7α-hydroxycholes-terol. A further step includes the activation of CYP8B1 for CA. CYP7A1 is involved in the synthesis of two primary BAs, CA and CDCA, and contributes to more than 75% of total BA production. The alternative pathway is initiated by sterol-27-hydroxylase (CYP27A1), which produces the intermediate 27-hydroxyc-holesterol and mainly CDCA. A further step includes the activation of CYP8B1 for CDCA. In the small and large intestine, the bacterial deconjugation, dehy-drogenation, 7α-dehydroxylation, and epimerization of the primary BAs produces the “secondary” BAs. CA is converted to the dihydroxy deoxychol ic acid (DCA) and CDCA to the monohydroxy lithocholic acid (LCA). The 7α-dehydrogenation of CDCA form the dihydroxy 7a-oxo-LCA which does not accumulate in bile, but is metabolized to a “tertiary” BA by hepatic or bacterial reduction to CDCA, mainly in the liver or its 7β-epimer, the dihydroxy ursodeoxycholic acid (UDCA), primarily by colonic bacteria.2,9 The position and orientation of the hydroxyls for each BA is indicated in parenthesis.2,9,59")
Major primary, secondary, and tertiary BAs of humans. The sites of BA synthesis and metabolism are shown. The “primary” BAs are synthetized in the liver from cholesterol as precursor.The trihydroxy cholic acid (CA) and the dihydroxy chenodeoxycholic acid (CDCA). Two bio synthetic pathways are involved: the classical pathway is initiated by 7α-hydroxylase (CYP7A1) which stimulates the 7α-hydroxylation of cholesterol with synthesis of 7α-hydroxycholes-terol. A further step includes the activation of CYP8B1 for CA. CYP7A1 is involved in the synthesis of two primary BAs, CA and CDCA, and contributes to more than 75% of total BA production. The alternative pathway is initiated by sterol-27-hydroxylase (CYP27A1), which produces the intermediate 27-hydroxyc-holesterol and mainly CDCA. A further step includes the activation of CYP8B1 for CDCA. In the small and large intestine, the bacterial deconjugation, dehy-drogenation, 7α-dehydroxylation, and epimerization of the primary BAs produces the “secondary” BAs. CA is converted to the dihydroxy deoxychol ic acid (DCA) and CDCA to the monohydroxy lithocholic acid (LCA). The 7α-dehydrogenation of CDCA form the dihydroxy 7a-oxo-LCA which does not accumulate in bile, but is metabolized to a “tertiary” BA by hepatic or bacterial reduction to CDCA, mainly in the liver or its 7β-epimer, the dihydroxy ursodeoxycholic acid (UDCA), primarily by colonic bacteria.2,9 The position and orientation of the hydroxyls for each BA is indicated in parenthesis.2,9,59
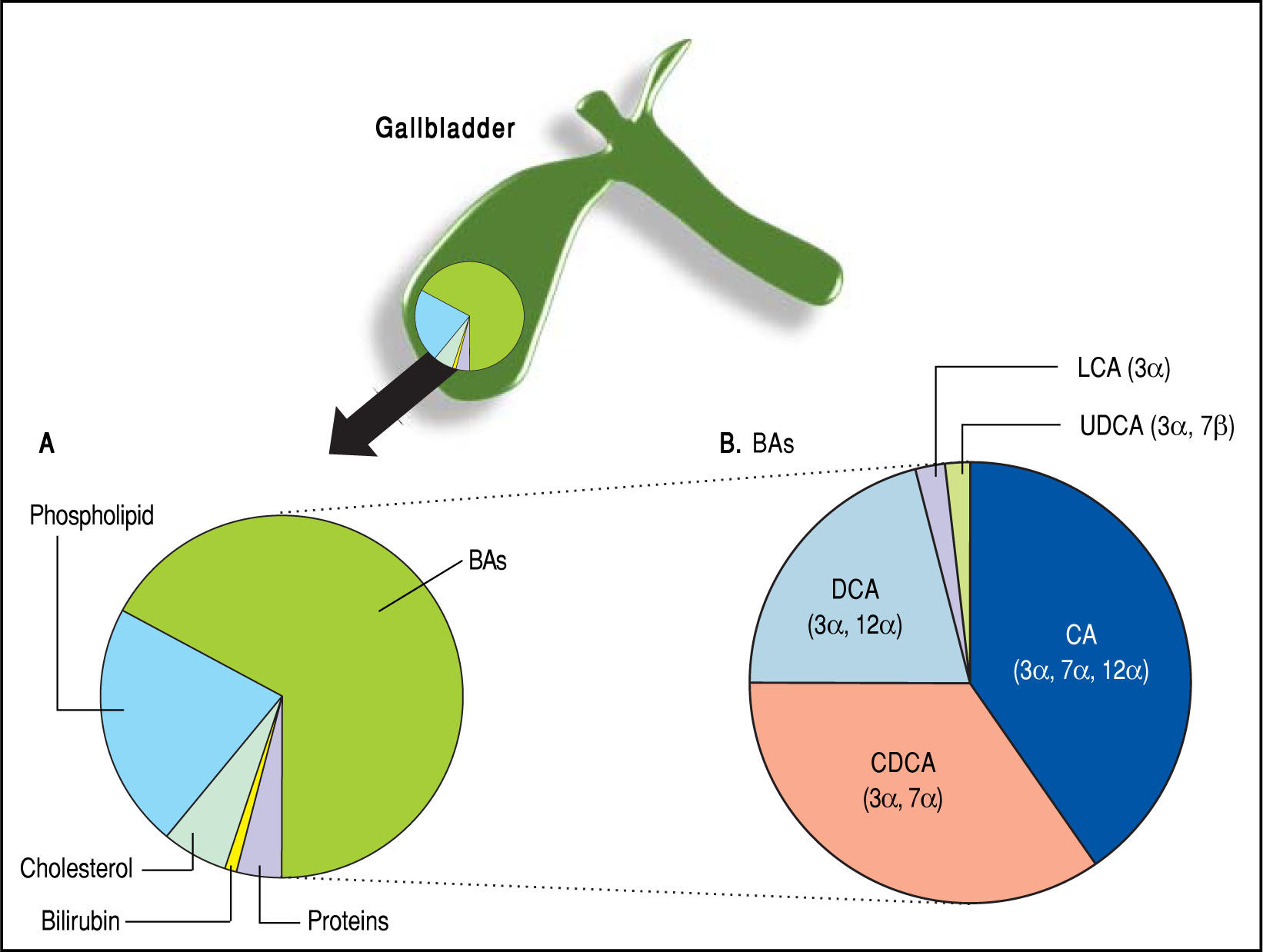
BAs constitute about two thirds of the solute mass of normal human bile by weight. They belong to the class of biliary lipids together with cholesterol and phospholi-pids (Figure 3). The complex scenario related to BA biosynthesis, enterohepatic circulation and interactions with ileal and liver receptors are depicted in Figure 4. Hepatic synthesis of BAs accounts for 0.2-0.6 g/day with an overall BA pool of about 3 g in the liver and intestine. More than 95% of the secreted BAs are reabsorbed through active absorption at the terminal ileum by the specific bile acid transporter apical sodium-dependent bile acid transporter (ASBT) and passive absorption in the colon. These BAs are recirculated to the liver via the portal vein, i.e., the so-called enterohepatic circulation, raising the overall pool to 3 g with the recirculation of 4-12 cycles per day, i.e., 12-36 g/day. Only about 5% (i.e., 0.2-0.6 g per day) of the secreted BAs are lost in feces, equal to the amount of hepatic synthesis (0.2-0.6 g/day).2

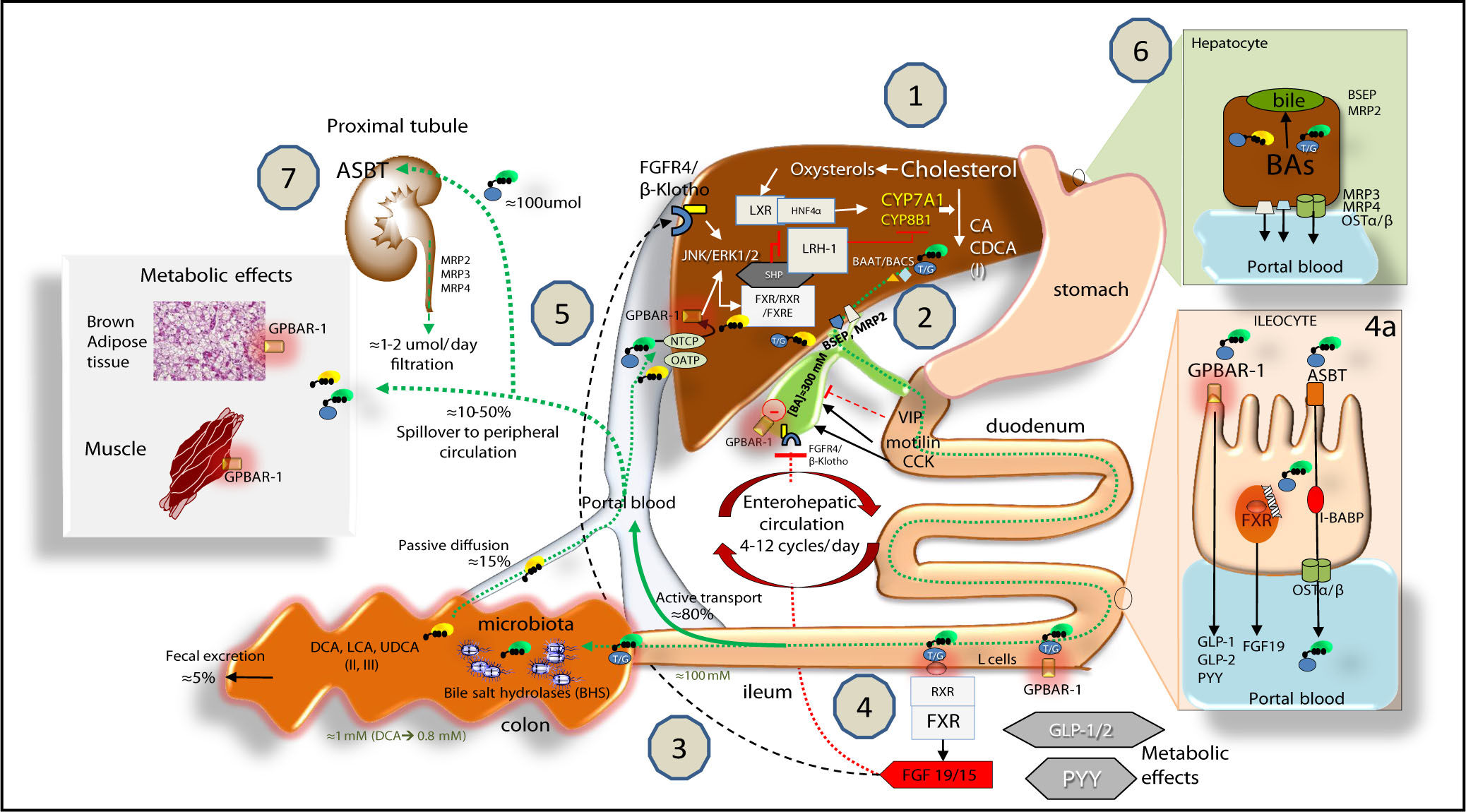
![Bile acid (BA) biosynthesis, enterohepatic circulation and function through their receptors in the liver and intestine.Complex molecular mechanisms involve a set of nuclear receptors, i.e., farnesoid X receptor (FXR), retinoid X receptor (RXR), small heterodimer partner (SHP), liver receptor homologous-1 (LRH-1), and liver X receptor (LXR).77 FXR plays a key role as main sensor of BAs and regulator of synthesis, secretion and metabolism of BAs in the liver, ileum and colon.78,791. In the liver, the primary BAs (CA, CDCA) are mainly synthesized from cholesterol by the rate-limiting microsomal enzyme cholesterol 7a-hydroxylase (CYP7A1) and by CYP8B1 at a later step (the “classical pathway”) and by the CYP27A1 (the “alternative pathway”). BAs are conjugated to taurine or glycine mainly via two enzymes, BA CoA synthase (BACS) and BA-CoA-amino acid N-acetyltransferase (BAAT), secreted into bile by the bile salt export pump (BSEP); the multidrug resistance-associated protein (MRP2) mediates secretion of organic substrates such as bilirubin, and glutathione. 2. Gallbladder: bile is stored and concentrated because water absorption occurs, as well as periodically released into the duodenum due to gallbladder contraction in the fasting state (about 20% emptying at the end of phase II of the migrating myoelectric complex80,81 under the control of the vagus and enterohormone motilin81 and especially after a meal due to the enterohormone cholecystokinin, CCK8). This rythmic activity is also modulated in concert with episodes of gallbladder relaxation/refilling due to the effect of the vasointestinal peptide (VIP, released in the duodenum by gastric acid), BAs per se (acting on the gallbladder receptor GPBAR-1), and the intestinal FGF15/19 (following the BA/FXR interaction in the ileum) acting on the FGF4/(l-Klotho receptor also expressed in the gallbladder.8,82,83 Fasting serum BAs concentrations in healthy subjects are 0.2-0.7 \M to increase to 4-5\M after each meal2-53. BAs are efficiently (i.e., > 95%) reabsorbed in the terminal ileum. The remaining BAs enter the colon, undergo biotransformation to the secondary BAs by the resident gut microbiota, and undergo passive diffusion and reabsorption. Only 5% of BAs are lost in feces every day. The enterohepatic circulation of BAs includes their intestinal re-absorption and continuous recirculation to the liver through the portal vein. About 10-50% of re-absorbed BAs undergo peripheral spillover into systemic circulation.844. Upon arrival in the terminal ileum, BAs activate FXR and increase the transcription of the enterokine fibroblast growth factor 19 (FGF19 in humans or FGF15 in mice) which enters the portal circulation and regulates both gallbladder (see point 2) and liver effects (see point 5). BAs in the intestine also activate the G protein-coupled receptor (GPBAR-1) and stimulate the secretion of peptide YY (PYY), glucagon-like peptide 1 (GLP-1) and glucagon-like peptide 2 (GLP-2), all of which produce important metabolic effects on glucose metabolism,85 insulin metabolism and appetite acting on GPBAR-1 receptors located in the cells of brown adipose tissue and muscle.85 In the ileocyte, BA uptake, intracellular transport and secretion into the portal vein require the apical sodium dependent bile acid transporter (ASBT), the cellular intestinal BA binding protein (I-BABP), and the basolateral heterodimeric organic solute transporter (OSTa/p), respectively (see inset 4a for details). 5. The circulating FGF19 binds to hepatic FGF receptor 4 (FGFR4)/(l-Klotho to activatec-Jun N-terminal kinase/extracellular signal-regulated kinase (JNK/ERK) signaling, which inhibits expression of CYP7A1 and CYP8B1 and hepatic BA synthesis, in synergy with the FXR-SHP inhibitory pathway.7086 BAs enter the liver by sodium taurocholate cotransporting polypeptide (NTCP) and organic anion transporting polypeptide (OATP) transporters and act as physiological nuclear ligands for FXR, which regulates target gene transcription by binding toRXRs as a heterodimer.87 This results in increased transcription of the small heterodimer partner (SHP) expression. SHP, in turn, inhibits LRH-1, preventing the activation of target genes that participate in BA and fatty acid synthesis. In the absence of BAs, LRH-1 acts together with LXR to stimulate BA synthesis.55,88,89 FXR also regulates the enzymatic activity that is involved in BA conjugation to glycine or taurine, and hepatic BA secretion by of BSEP and hepatic phospholipid secretion by ABCB4. BAs re-entering the liver also interact with the liver GPBAR-1 expressed in Kupffer cells, in concert with the pathway activated by the FGFR4/(l-Klotho. FXR activation also coordinates BA detoxification enzymes (i.e., cytosolic sulfotransferase 2A1 [SULT2A1], aldol-keto reductase 1 B7 [AKR1B7], cytochrome P450 3A4/3a11 [CYP3A4/ Cyp3a11], and UDP-glycosyltransferase 2B4 [UTG2B4]).906. The events leading to BA excretion from the hepatocyte into the portal vein are shown in the inset. Specific transporters are the multidrug resistance protein 3 and 4 (MRP3, MRP4) and OSTa/p. 7. From the peripheral circulation, BAs also undergo renal uptake by the apical sodium/dependent bile acid transporter (ASBT) in the proximal tubule. Glomerular filtration of BAs are regulated by MRP2, 3, 4 transporters.91 Adapted from Ory, et al.92 and Inagaki, et al.,70 Garruti, et al.,77 Liu, et al.52](https://static.elsevier.es/multimedia/16652681/00000016000000S1/v1_201906111104/S1665268119310385/v1_201906111104/en/main.assets/gr4.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNeKOJqKH5+Za6P+RA5Vq9Dlpy3cNuM9SlG6iEvGuhjp1TcS3Nc+k7oSysI41+1NG7Hj447cXH5ZfQc2e0Hm3dpidRJ17kKS7LxKZNfsq1J1cgjzOjSNRfkolRX8yFruC18BExxlWwIh9HRMAGoJBftwum3YeIsaskqm7jB09BWXgRSGhQanRuWevHjjTO4tC6fluEdEawU5D0TU0fKqcHRWCpcUfz20QVvJwzwTEwEfOl67nFyNMOilJqpvy/HQ/XA= "Bile acid (BA) biosynthesis, enterohepatic circulation and function through their receptors in the liver and intestine.Complex molecular mechanisms involve a set of nuclear receptors, i.e., farnesoid X receptor (FXR), retinoid X receptor (RXR), small heterodimer partner (SHP), liver receptor homologous-1 (LRH-1), and liver X receptor (LXR).77 FXR plays a key role as main sensor of BAs and regulator of synthesis, secretion and metabolism of BAs in the liver, ileum and colon.78,791. In the liver, the primary BAs (CA, CDCA) are mainly synthesized from cholesterol by the rate-limiting microsomal enzyme cholesterol 7a-hydroxylase (CYP7A1) and by CYP8B1 at a later step (the “classical pathway”) and by the CYP27A1 (the “alternative pathway”). BAs are conjugated to taurine or glycine mainly via two enzymes, BA CoA synthase (BACS) and BA-CoA-amino acid N-acetyltransferase (BAAT), secreted into bile by the bile salt export pump (BSEP); the multidrug resistance-associated protein (MRP2) mediates secretion of organic substrates such as bilirubin, and glutathione. 2. Gallbladder: bile is stored and concentrated because water absorption occurs, as well as periodically released into the duodenum due to gallbladder contraction in the fasting state (about 20% emptying at the end of phase II of the migrating myoelectric complex80,81 under the control of the vagus and enterohormone motilin81 and especially after a meal due to the enterohormone cholecystokinin, CCK8). This rythmic activity is also modulated in concert with episodes of gallbladder relaxation/refilling due to the effect of the vasointestinal peptide (VIP, released in the duodenum by gastric acid), BAs per se (acting on the gallbladder receptor GPBAR-1), and the intestinal FGF15/19 (following the BA/FXR interaction in the ileum) acting on the FGF4/(l-Klotho receptor also expressed in the gallbladder.8,82,83 Fasting serum BAs concentrations in healthy subjects are 0.2-0.7 \M to increase to 4-5\M after each meal2-53. BAs are efficiently (i.e., > 95%) reabsorbed in the terminal ileum. The remaining BAs enter the colon, undergo biotransformation to the secondary BAs by the resident gut microbiota, and undergo passive diffusion and reabsorption. Only 5% of BAs are lost in feces every day. The enterohepatic circulation of BAs includes their intestinal re-absorption and continuous recirculation to the liver through the portal vein. About 10-50% of re-absorbed BAs undergo peripheral spillover into systemic circulation.844. Upon arrival in the terminal ileum, BAs activate FXR and increase the transcription of the enterokine fibroblast growth factor 19 (FGF19 in humans or FGF15 in mice) which enters the portal circulation and regulates both gallbladder (see point 2) and liver effects (see point 5). BAs in the intestine also activate the G protein-coupled receptor (GPBAR-1) and stimulate the secretion of peptide YY (PYY), glucagon-like peptide 1 (GLP-1) and glucagon-like peptide 2 (GLP-2), all of which produce important metabolic effects on glucose metabolism,85 insulin metabolism and appetite acting on GPBAR-1 receptors located in the cells of brown adipose tissue and muscle.85 In the ileocyte, BA uptake, intracellular transport and secretion into the portal vein require the apical sodium dependent bile acid transporter (ASBT), the cellular intestinal BA binding protein (I-BABP), and the basolateral heterodimeric organic solute transporter (OSTa/p), respectively (see inset 4a for details). 5. The circulating FGF19 binds to hepatic FGF receptor 4 (FGFR4)/(l-Klotho to activatec-Jun N-terminal kinase/extracellular signal-regulated kinase (JNK/ERK) signaling, which inhibits expression of CYP7A1 and CYP8B1 and hepatic BA synthesis, in synergy with the FXR-SHP inhibitory pathway.7086 BAs enter the liver by sodium taurocholate cotransporting polypeptide (NTCP) and organic anion transporting polypeptide (OATP) transporters and act as physiological nuclear ligands for FXR, which regulates target gene transcription by binding toRXRs as a heterodimer.87 This results in increased transcription of the small heterodimer partner (SHP) expression. SHP, in turn, inhibits LRH-1, preventing the activation of target genes that participate in BA and fatty acid synthesis. In the absence of BAs, LRH-1 acts together with LXR to stimulate BA synthesis.55,88,89 FXR also regulates the enzymatic activity that is involved in BA conjugation to glycine or taurine, and hepatic BA secretion by of BSEP and hepatic phospholipid secretion by ABCB4. BAs re-entering the liver also interact with the liver GPBAR-1 expressed in Kupffer cells, in concert with the pathway activated by the FGFR4/(l-Klotho. FXR activation also coordinates BA detoxification enzymes (i.e., cytosolic sulfotransferase 2A1 [SULT2A1], aldol-keto reductase 1 B7 [AKR1B7], cytochrome P450 3A4/3a11 [CYP3A4/ Cyp3a11], and UDP-glycosyltransferase 2B4 [UTG2B4]).906. The events leading to BA excretion from the hepatocyte into the portal vein are shown in the inset. Specific transporters are the multidrug resistance protein 3 and 4 (MRP3, MRP4) and OSTa/p. 7. From the peripheral circulation, BAs also undergo renal uptake by the apical sodium/dependent bile acid transporter (ASBT) in the proximal tubule. Glomerular filtration of BAs are regulated by MRP2, 3, 4 transporters.91 Adapted from Ory, et al.92 and Inagaki, et al.,70 Garruti, et al.,77 Liu, et al.52")
Bile acid (BA) biosynthesis, enterohepatic circulation and function through their receptors in the liver and intestine.Complex molecular mechanisms involve a set of nuclear receptors, i.e., farnesoid X receptor (FXR), retinoid X receptor (RXR), small heterodimer partner (SHP), liver receptor homologous-1 (LRH-1), and liver X receptor (LXR).77 FXR plays a key role as main sensor of BAs and regulator of synthesis, secretion and metabolism of BAs in the liver, ileum and colon.78,791. In the liver, the primary BAs (CA, CDCA) are mainly synthesized from cholesterol by the rate-limiting microsomal enzyme cholesterol 7a-hydroxylase (CYP7A1) and by CYP8B1 at a later step (the “classical pathway”) and by the CYP27A1 (the “alternative pathway”). BAs are conjugated to taurine or glycine mainly via two enzymes, BA CoA synthase (BACS) and BA-CoA-amino acid N-acetyltransferase (BAAT), secreted into bile by the bile salt export pump (BSEP); the multidrug resistance-associated protein (MRP2) mediates secretion of organic substrates such as bilirubin, and glutathione. 2. Gallbladder: bile is stored and concentrated because water absorption occurs, as well as periodically released into the duodenum due to gallbladder contraction in the fasting state (about 20% emptying at the end of phase II of the migrating myoelectric complex80,81 under the control of the vagus and enterohormone motilin81 and especially after a meal due to the enterohormone cholecystokinin, CCK8). This rythmic activity is also modulated in concert with episodes of gallbladder relaxation/refilling due to the effect of the vasointestinal peptide (VIP, released in the duodenum by gastric acid), BAs per se (acting on the gallbladder receptor GPBAR-1), and the intestinal FGF15/19 (following the BA/FXR interaction in the ileum) acting on the FGF4/(l-Klotho receptor also expressed in the gallbladder.8,82,83 Fasting serum BAs concentrations in healthy subjects are 0.2-0.7 \M to increase to 4-5\M after each meal2-53. BAs are efficiently (i.e., > 95%) reabsorbed in the terminal ileum. The remaining BAs enter the colon, undergo biotransformation to the secondary BAs by the resident gut microbiota, and undergo passive diffusion and reabsorption. Only 5% of BAs are lost in feces every day. The enterohepatic circulation of BAs includes their intestinal re-absorption and continuous recirculation to the liver through the portal vein. About 10-50% of re-absorbed BAs undergo peripheral spillover into systemic circulation.844. Upon arrival in the terminal ileum, BAs activate FXR and increase the transcription of the enterokine fibroblast growth factor 19 (FGF19 in humans or FGF15 in mice) which enters the portal circulation and regulates both gallbladder (see point 2) and liver effects (see point 5). BAs in the intestine also activate the G protein-coupled receptor (GPBAR-1) and stimulate the secretion of peptide YY (PYY), glucagon-like peptide 1 (GLP-1) and glucagon-like peptide 2 (GLP-2), all of which produce important metabolic effects on glucose metabolism,85 insulin metabolism and appetite acting on GPBAR-1 receptors located in the cells of brown adipose tissue and muscle.85 In the ileocyte, BA uptake, intracellular transport and secretion into the portal vein require the apical sodium dependent bile acid transporter (ASBT), the cellular intestinal BA binding protein (I-BABP), and the basolateral heterodimeric organic solute transporter (OSTa/p), respectively (see inset 4a for details). 5. The circulating FGF19 binds to hepatic FGF receptor 4 (FGFR4)/(l-Klotho to activatec-Jun N-terminal kinase/extracellular signal-regulated kinase (JNK/ERK) signaling, which inhibits expression of CYP7A1 and CYP8B1 and hepatic BA synthesis, in synergy with the FXR-SHP inhibitory pathway.7086 BAs enter the liver by sodium taurocholate cotransporting polypeptide (NTCP) and organic anion transporting polypeptide (OATP) transporters and act as physiological nuclear ligands for FXR, which regulates target gene transcription by binding toRXRs as a heterodimer.87 This results in increased transcription of the small heterodimer partner (SHP) expression. SHP, in turn, inhibits LRH-1, preventing the activation of target genes that participate in BA and fatty acid synthesis. In the absence of BAs, LRH-1 acts together with LXR to stimulate BA synthesis.55,88,89 FXR also regulates the enzymatic activity that is involved in BA conjugation to glycine or taurine, and hepatic BA secretion by of BSEP and hepatic phospholipid secretion by ABCB4. BAs re-entering the liver also interact with the liver GPBAR-1 expressed in Kupffer cells, in concert with the pathway activated by the FGFR4/(l-Klotho. FXR activation also coordinates BA detoxification enzymes (i.e., cytosolic sulfotransferase 2A1 [SULT2A1], aldol-keto reductase 1 B7 [AKR1B7], cytochrome P450 3A4/3a11 [CYP3A4/ Cyp3a11], and UDP-glycosyltransferase 2B4 [UTG2B4]).906. The events leading to BA excretion from the hepatocyte into the portal vein are shown in the inset. Specific transporters are the multidrug resistance protein 3 and 4 (MRP3, MRP4) and OSTa/p. 7. From the peripheral circulation, BAs also undergo renal uptake by the apical sodium/dependent bile acid transporter (ASBT) in the proximal tubule. Glomerular filtration of BAs are regulated by MRP2, 3, 4 transporters.91 Adapted from Ory, et al.92 and Inagaki, et al.,70 Garruti, et al.,77 Liu, et al.52
Most of BAs remain in the enterohepatic circulation and only minimum quantity enters the blood circulation and is excreted into urine by the kidneys (< 1 μM/day). However, biliary secretion of BAs is compromised in hepatobiliary diseases. To alleviate BA accumulation, BAs undergo sulfation by the enzyme sulfotransferase 2A1 (SULT2A1), mainly in the liver. The sulfated BAs are more water soluble, and consequently, their absorption rates in the intestines are decreased, while urinary elimination is increased over 100 times under these condi-tions.10 The sulfated BAs constitute over 89% of urinary BAs, and the majority of them are also amidated with gly-cine or taurine. The degree of sulfation of BAs is inversely related to their hydrophobicity, with LCA being almost entirely sulfated in urine, while only half of the CA is found to be sulfated. These results point to a critical role for this detoxifying mechanism.11
The major physiological functions of BAs include the digestion and absorption of intestinal cholesterol, triglyc-erides, fatty acids,12 and fat-soluble vitamins,13 feedback regulatory mechanisms of hepatic BA biosynthesis, and gallbladder motor function.8 BAs also play a critical role in the gut-liver axis in response to inflammation,14 immune response,15-17 epithelial cell proliferation,18 intestinal microbiota19 and gene expression through epigenetic mechanisms.20 Recently, a new role of BAs has been proposed for regulating transintestinal cholesterol excretion and reverse cholesterol transport.21
Bas and Physical States of Biliary LipidsBAs tend to self-assemble into micelles in an aqueous solution when the critical micellar concentration (CMC) is exceeded. Normally, the CMC values of most of BAs are between 1 and 20 μM, but this value is greatly dependent on the species of BAs, the ionic strength and composition, and types and concentrations of other solubilized lipids. Another factor which influences the CMC is the progressive bile concentration within the biliary tree and especially in the gallbladder, in such a way that BA concentration steadily exceeds the CMCs.2 As a result, simple micelles are formed in bile, which are able to solubilize other types of lipids such as cholesterol and phospholip-ids and lead to the formation of mixed micelles in bile.
Cholesterol and lecithins are virtually insoluble in water, and bile is an aqueous solution. Thus, cholesterol and lecithins require BAs for their transport in bile because BAs contain both hydrophilic and hydrophobic areas, which confer the property of amphiphilicity. The number and characteristics of hydroxyl groups and side chains characterize their solubility in water and bile according to the composition and concentration of other lipids. Thus, BA aggregation in bile leads to a transformation from monomers to simple micelles if concentration exceeds the CMCs (~ 2 mmol/L). The simple micelles can solubilize cholesterol because the hydrophobic portion of each BA molecule faces in ward and the hydrophilic groups go outward, with cholesterol being solubilized within the central hydrophobic portion of the micelle. BA simple micelles appear like disks, ~ 3 nm in diameter. After the incorporation of phospholipids into simple micelles, mixed micelles are formed and they are ~ 4-8 nm in diameter, with the capacity to solubilize 3 times more cholesterol. The mixed (BA-cholesterol-phospholipid) micelles appear as a lipid bilayer. The hydrophilic groups of the BAs and phospholipids are on the “outside” of the bilayer in contact with the aqueous bile, and cholesterol molecules are often solubilized by the hydrophobic groups on the “inside” of the bilayer. The maximal solubility of cholesterol occurs when the molar ratio of phospholipids to BAs is between 0.2 and 0.3, and more cholesterol is solu-bilized when the concentration of total biliary lipids increases.
Using quasi-elastic light-scattering spectroscopy and electron microscopy, model and native human bile is studied22-24 to depict the pathways of biliary cholesterol solubilization, which involve the formation of biliary unilamellar vesicles25,26 and liquid crystals(i.e., multila-mellar vesicles)27,28 as well as cholesterol nucleation and crystallization in bile.29 The size of unilamellar vesicles is ~ 40 to 100 nm in diameter, and they are spherical structures with a single bilayer that encircles an aqueous core. These structures are enriched with phospholipids and cholesterol, but little BAs. Unilamellar vesicles can aggregate and fuse to form large multilamellar vesicles (liquid crystals or liposomes, ~ 500 nm in diameter), which consist of multilamellar concentric spherical structures. These vesicles are able to solubilize biliary cholesterol that cannot be solubilized in simple and mixed micelles. The compositions and proportions of micelles and vesicles depend on the concentrations of biliary lipids: with a dilute bile (i.e., total lipid concentration < 3 g/dL), vesicles are stable (i.e.,no aggrega-tionor fusion, or nucleation of solid cholesterol crystals). For concentrated gallbladder bile (~ 10 g/dL), vesicle instability is significantly increased, leading to the precipitation of solid cholesterol crystals. In the fasting state, hepatic BA output is relatively low and cholesterol is carried more in vesicles than in micelles. During meals, the BA output is higher, and therefore, more cholesterol is solubilized in micelles. With increasing BA concentrations (i.e., in concentrated gallbladder bile), more multilamellar vesicles form. If the ratio of cholesterol to phospholipid in vesicles is greater than 1, vesicles become increasingly unstable, which could lead to the formation of solid plate-like cholesterol monohy-drate crystals, the first step in cholesterol nucleation. The following events leading to the formation of solid cholesterol crystals and gallstone formation have been summarized by our group in previous papers.2,30-32
Bas and the MicrobiotaThe maintenance of an appropriate BA pool in the body is determined by hepatic BA synthesis, biliary secretion, gallbladder concentration and contraction, intestinal transit, microbial biotransformation, intestinal re-absorption and fecal excretion (Figure 4). Intestinal bacteria play a major role in BA metabolism because they are responsible for the transformation of the primary BAs to the secondary BAs. The involved steps include deconjugation, oxidation of hydroxyl groups in 3, 7 and 12 positions, and 7-dehydroxylation.6 The human gut microbiota is an extremely dynamic system in both health and disease,33-35 and therefore, has profound effects on the final BA profile. This process significantly increases the hydrophobicity of the BA pool, and as a consequence, the risk of potential carcinogenic effects.36
On the other hand, BAs (mainly DCA) have antimicrobial properties and are able to influence the species of gut microbiota. This is done by the detergent effects of DCA on bacterial cell membranes, which damages the integrity of the bacteria and modulates microbial populations.19 The obstruction of bile flow is a major factor for bacterial overgrowth and translocations in the intestine. In a mouse model, this condition is reversed by oral administration of BAs. This effect is modulated by FXR-induced gene expression, which is associated with the enteral protection and the inhibition of bacteria damage to the intestinal mu-cosa.37
Furthermore, the gut microbiota can be considered a privileged interface between the environment (including dietary habits at high risk for cancer development,38-40 smoking,41 ethanol consumption,42 environmental pollutants as heavy metals and pesticides43-46) and BAs-mediated signaling pathways7 regulating intestinal and metabolic homeostasis and potentially inducing cancer onset and growth.36
Bas and Dietary HabitsDiets containing high content of animal proteins and saturated fats increase bile secretion, augmenting BAs in the intestine. These alterations markedly influence the gut microbiota40,47,48 by favoring bacteria to increase the concentration of hydrophobic BAs (mainly DCA47,48) in the total BA pool.39,49 In mice, a high-fat diet decreases Lactobacillales and increases the Clostridium subcluster XIVa, leading to an increase in serum levels of DCA. The modified microbiota composition is suppressed by dietary supplement of agaro-oligosaccharides (a natural derivate from agarose).48 In Apcmin/+ mice, treatment with DCA alters the gut microbiota composition by causing defective intestinal barrier function, intestinal low-grade inflammation, and cancer progression. When fecal microbiota is transplanted from DCA-treated mice to another group of Apcmin/+ animals, an increased tumor multiplicity is found likely due to activation of the tumor-associated Wnt/β-catenin signaling pathway. Of note, the cancer-promoting effects of BAs are blocked by gut microbiota depletion through antibiotic treatment.36 As demonstrated in animals, the pathways linking a high-fat diet to an alteration in intestinal microbiota are also correlated with increased retention of hydrophobic BAs in the liver, leading to hepatocellular carcinoma in animals with nonalcoholic steatohepatitis (NASH). This pathogenic mechanism isalso supported by an inhibition of key BA transporters secondary to high-fat diet-induced liver inflammation and to a down-regulation of the tumor suppressor gene CEBPα.50
Bas as Signaling MoleculesAs mentioned above, BAs display both hydrophilic and hydrophobic surfaces, which makes these molecules highly soluble, and detergent-like amphiphilic. The potency of BAs as detergents depends on the distribution and orientation of hydroxyl groups around the steroid nucleus of the molecule, a feature called hydrophobicity, which is quantified by high performance liquid chromatography (HPLC).2 The hydrophobicity of BAs, which is directly related to cytotoxicity, is the following order: LCA> DCA > CDCA > CA > UDCA.
BAs are also being recognized as signaling molecules in the human body because they are able to regulate metabolic and cellular functions by interaction with BA receptors. BAs interact with the nuclear receptor superfamily such as ligand-activated FXR and GPBAR-1.1 FXR is a master BA sensor in the liver and ileum.51-53 The BA-FXR interaction is essential in BA homeostasis: the rank order of potency is CDCA > LCA = DCA > CA in the conjugated and un-conjugated forms.54 FXR regulates a series of gene expression that is involved in the synthesis, uptake, secretion and intestinal absorption of BAs, and all these processes are essential in the regulation of intracellular Bas.2,55,56 FXR activation in the intestine increases expression of intestinal fibroblast growth factor 19 (i.e., FGF19 in humans or FGF15 in mice); in turn, the circulating FGF19 enters the liver via the portal vein and reduces expression of hepatic cholesterol 7α-hydroxylase and BA synthesis.57
BAs also interact with GPBAR-1 that is mainly expressed in Kupffer cells, but not hepatocytes.39,58 In this case, the rank order of potency is TLCA > TDCA > TCDCA > TCA.1 In the ileum, activation of GPBAR-1 increases levels of peptide YY (PYY) with anorexigenic effect (i.e., appetite reduction), as well as glucagon-like pep-tide-1 (GLP-1) and glucagon-like peptide-2 (GLP-2).60 GPBAR-1 is also expressed and metabolically active in the gallbladder, brown adipose tissue, skeletal muscle, macro-phages, and monocytes1,59 and in the enteroendocrine cells of the intestine.61 In particular, GPBAR-1 signalling in skeletal muscle and brown adipose tissue results in local activation of the type II iodothyronine deiodinase (DIO2) able to generate or transform the inactive thyroxine (T4) to active thyroid hormone (T3, a key regulator of metabolism and energy homeostasis). In Kupffer cells and macro-phages, GPBAR-1 activation inhibits LPS-induced cytokine production.62 Such additional hormonal effects of BAs are cAMP-mediated and might be particularly evident after bariatric surgery with important and beneficial metabolic effects, including increased energy expenditure, increased insulin secretion and/or sensitivity and decrease inflammatory status.9,57,62 (see also chapter by Garruti, et al. in the present issue). The mechanisms governing BA biosynthesis and the composition of the total BA pool are, therefore, of paramount importance for keeping the overall digestive and metabolic functions of BAs in health. This aspect involves a number of nuclear receptors in the liver and intestine via FXR-dependent and -independent mechanisms.
Fxr-Dependent MechanismsOverall, cholesterol 7α-hydroxylase (CYP7A1) is the rate-limiting enzyme for regulating BA synthesis and is a target gene of FXR. Several factors such as BAs, inflammatory cytokines, steroid hormones, and insulin may inhibit CYP7A1 transcription through the 5’-upstream region of the promoter.63-65 FXR is critical in this respect as a regulatory factor of BA metabolism because it down-regulates CYP7A1, sterol 12α-hydroxylase (CYP8B1), and sterol 27-hydroxylase (CYP27A1) transcription by a negative feedback mechanism.
In the hepatocyte, one regulatory mechanism involves binding of BAs to FXR in the nucleus, the formation of the FXR/RXR heterodimer, and the activationof small heterodimer partner (SHP), leading to an inhibition of the activity of liver receptor homol-ogous-1 (LRH-1) and the CYP7A1 transcription.55,66 Furthermore, SHP displaces the promoter factor HNF4α from PGC-1α, thus contributing to CYP7A1 and CYP8B1 transcription. FXR also plays a role in decreasing BA cytotoxicity because it promotes expression of the enzymes involved in conjugation of BAs with glycine or taurine, i.e., BA CoA synthase and BA-CoA-amino acid N-acetyltransferase.67,68 Excessive accumulation of intrahepatic triglycerides during the sequence non-alcoholic fatty liver, steatohepatitis is associated with abnormalities of gene expression of FXR and SHP and BA transporters.69
In the ileum, a second regulatory mechanism of BA synthesis involves the secretion of FGF19 and activation of FGFR4 tyrosine kinase/β-klotho (a co-expressed membrane-bound glycosidase) signaling in the hepatocyte ba-solateral membrane.70,71 This pathway involves the JNK-mediated pathway and suppression of CYP7A1 transcription and points to the importance of the BAs/FXR/ FGF19/FGFR4/CYP7A1 signaling cascade which negatively regulates BA biosynthesis in the liver in hu-mans.2,72,73
FXR-independent mechanismsFXR-independent BA inhibition of CYP7A1 transcription might work by several parallel mechanisms to protect against BA toxicity during cholestasis and liver injury.
- •
Insulin receptor and activation of PI3K and AKT lead to phosphorylation of FoxO1 and inhibit CYP7A1 transcription.
- •
Activation of the pregnane X receptor (PXR) and vitamin D receptor by LCA and binding to the BA response element (BARE)-I sequence in the CYP7A1 promoter may inhibit CYP7A1 promoter activity.74
- •
Also, both PXR and vitamin D receptor inhibit CYP7A1 transcription by blocking HNF4α recruitment of PGC-1α to CYP7A1 chromatin.
- •
BAs also activate epidermal growth factor receptor (EGFR) and the Raf-1/MEK/ERK signaling pathway, thus inhibiting CYP7A1 transcription.
- •
The hepatocyte growth factor (HGF) is released from hepatic stellate cells during liver regeneration and injury, and HGF stimulates HGF receptor cMet and MAPK pathways, leading to inhibition of CYP7A1 transcription and BA synthesis.
- •
Kupffer cells secrete TGFβ-1 that activate its receptor TRβII and the SMAD signaling pathway in the hepato-cyte. SMAD3 enters the nucleus of hepatocyte and works with HDACs and mSin3A to inhibit HNF4α activation of CYP7A1 transcription. A tumour suppressor p53 interacts with HNF4α and inhibits HNF4α activity. These alterations may inhibit CYP7A1 transcription.
- •
Under certain circumstances, i.e., endotoxin-induced cholestasis, lipopolysaccharides released by bacteria stimulate the secretion of TNFα (alpha) and IL-1β from Kupffer cells, leading to activation of Toll-like receptor 4. TNFα and IL-1β may inhibit CYP7A1 transcription by activating the TNF-α receptor and the MAPK/JNK pathway in the hepatocyte. JNK may inhibit CYP7A1 and CYP8B1 transcription and BA synthesis by phosphorylating cJun no period75,76 and HNF4α.
BAs are synthesized mainly in the liver and are the major lipid components of bile, as well as involved in hepatic cholesterol catabolism. BAs released by the gallbladder enter the gastrointestinal tract during the meal and are key regulators of fat emulsion and solubilisation, two essential steps for the digestion and absorption of cholesterol, triglycerides and fat-soluble vitamins. BAs also act as signaling molecules by activating two main sensors in the body: the nuclear receptor FXR and the cell surface receptor GPBAR-1. In this way, BAs become key regulators of complex homeostatic pathways at a systemic level ranging from their own homeostasis tocholesterol, triglyceride, glucose and energy metabolisms. Additional regulations include cell proliferation, inflammation, and tumor onset and progression.
Thus, maintaining the precise balance between BA species and amounts, as well as preventing the accumulation of excessive BAs in the body are of importance under-physiological or pathophysiological conditions that involve the liver, intestine, muscle and adipose tissues.
In addition, the pathways involving the intestinal microbiota and epigenetic factors regulate gene expression and act as a common interface between environmental factors (including diet, lifestyle, and exposure to environmental toxics) and the molecular events promoting the onset and the progress of disease. The high-fat diet, for example, increases the fecal concentration of the secondary BAs that are a risk factor for the development of colorectal cancer. Of note, intestinal microbiota and the epigenome might be modulated by the primary prevention strategies (i.e., changes in dietary habits and lifestyle, and reduced exposure to environmental toxics) and therapeutic tools. Future studies are needed to better clarify how these measures could influence pathogenic mechanisms, disease onset and the efficacy of the available therapeutic tools.
Abbreviations- •
BAs: bile acids
- •
CA: cholic acid
- •
CDCA: chenodeoxycholic acid
- •
CMC: critical micellar concentration
- •
CYP7A1: cholesterol 7á-hydroxylase
- •
DCA: deoxycholic acid
- •
FGF: fibroblast growth factor
- •
FXR: farnesoid X receptor
- •
GPBAR-1: G-protein-coupled bile acid receptor-1 (also known as TGR5)
- •
JNK: c-Jun N-terminal kinase
- •
LCA: lithocholic acid
- •
UDCA: ursodeoxycholic acid
We declare that we have no conflicts of interest.
AcknowledgementsThe present chapter is written in the context of the project FOIE GRAS, which has received funding from the European Union’s Horizon 2020 Research and Innovation programme under the Marie Sklodowska-Curie Grant Agreement No. 722619. Raquel Lunardi Baccetto and Emilio Molina-Molina are recipients of Foie Gras Early Research Training Grant.



