



Hepatic encephalopathy and brain edema are important complications in the course of a patient with acute liver failure. Presumed unrelated for many years, increasing evidence suggests that an increase in brain water is seen in all forms of hepatic encephalopathy. Ammonia, traditionally linked to the pathogenesis of hepatic encephalopathy, plays an important role in the increase in brain water. In acute liver failure, an osmotic disturbance in the astrocyte, in combination with an alteration of cerebral blood flow results in overt brain edema and intracranial hypertension. In cirrhosis, magnetic resonance techniques indicate the presence of a brain osmotic disturbance. Several clinical factors modulate the development of brain swelling.
Abbreviations:
Hepatic encephalopathy (HE)
Acute liver failure (ALF)
Portacaval anastomosis (PCA)
Cerebral blood flow (CBF)
Glial fibrillary acidic protein (GFAP).
Supported by a Merit Review from the VA Research Administration and the Stephen B. Tips Memorial Fund at Northwestern Memorial Hospital. Dr. Vaquero is supported by Fondo de Investigación Sanitaria (BEFI/FIS), Madrid, Spain.
Hepatic encephalopathy (HE) is a frequent clinical complication of patients with acute and chronic liver disease, and is defined as the presence of potentially reversible cerebral dysfunction in the absence of other known causes of brain disease.1 Brain edema is defined as the excessive accumulation of intracellular and/or extracellular fluid in brain tissue, and is a deadly complication of patients with acute liver failure (ALF) in deep HE.2 The development of these complications is an important event in the patient with liver disease. In cirrhosis, a first episode of encephalopathy signals a decreased survival and raises the need to evaluate liver transplantation in the appropriate candidate.3 In fulminant hepatic failure, the grade of hepatic encephalopathy is a strong predictor of outcome.4-6 Brain edema and intracranial hypertension remain a leading cause of death in ALF,4 despite important advances in intensive medical care.
This review highlights links between HE and brain edema, as well as discuss current concepts of the pathogenesis and pathophysiology of both complications. A clinical perspective is emphasized.
Brain edema and intracranial hypertension in ALFBrain edema and intracranial hypertension are more common in patients with a fulminant presentation of ALF8(Figure 1), while their frequency decrease in subacute cases. Conceptually, it is important to distinguish brain edema from intracranial hypertension. Even though brain edema leads to intracranial hypertension, the latter will have effects per se in the brain that may or may not coincide with those that cause brain edema. For this reason, the investigation of the pathogenesis of brain edema has been frequently hampered by the presence of various degrees of intracranial hypertension in different studies.

The cranium is a space with a fixed, non-distensible volume. Three different compartments are traditionally considered, with the brain parenchyma being the largest (approximately 80-90% of intracranial volume in normal subjects), followed by intravascular (approximately 10%) and cerebrospinal fluid (approximately 3-5%) compartments. Cells constitute most of the volume of brain parenchyma, with the extracellular fluid accounting just for 10-20% of that volume. This distribution is important as small increases in cell volume will lead to large increases in the total volume of the brain.
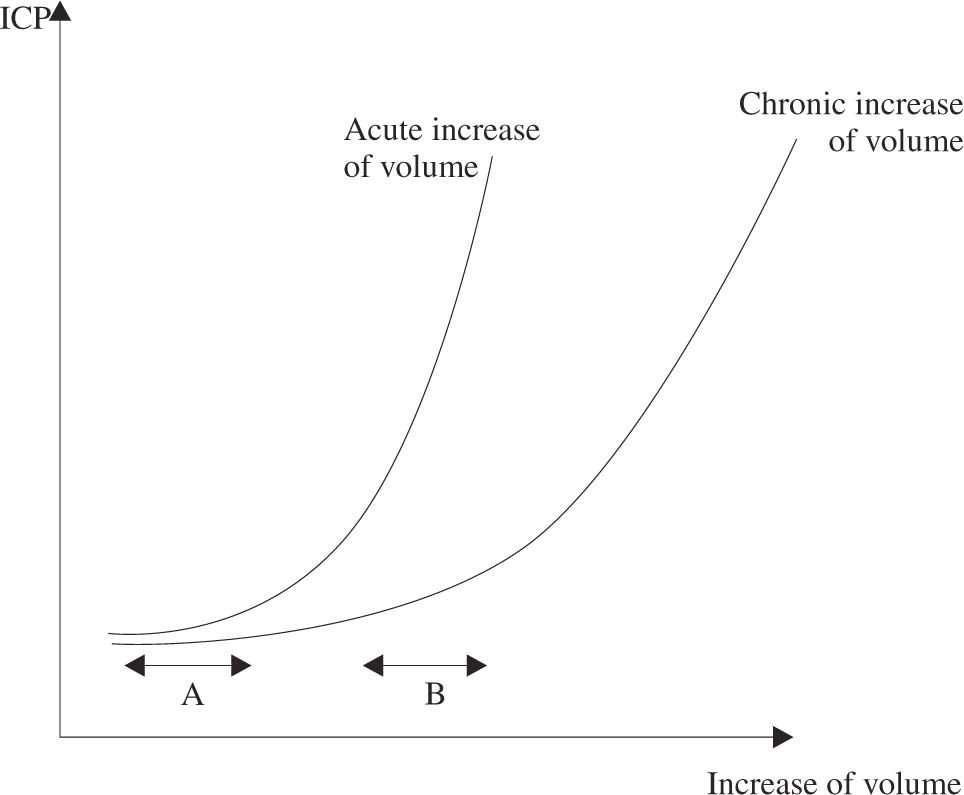
Intracranial hypertension is the result of an increase in volume in one or more of the previous compartments, without equivalent reduction of the others (Monro-Kelly doctrine). The capacity of the brain to compensate an increase in volume without a corresponding increase of intracranial pressure is called Compliance (ΔV/ΔP). Brain compliance has several important qualities (see figure 2). First, compliance is lower when the increase in volume is acute, which can leave no time for compensating mechanisms to develop. Second, when the increase in volume is continuous, compliance decreases progressively with time, leading ultimately to increased intracranial pressure. Third, when compliance is low, a small increase in volume, for example of cerebral blood volume, can lead to large increases of intracranial pressure.
. Continuous increases of volume leads to progressively larger increases of intracranial pressure. Even though the increase of volume is the same in A and B, the lower brain compliance results in higher increases of pressure in B. The curve will be steeper when the insult is acute, due to a lesser capacity of compensation.")
Relation between increase of volume and intracranial pressure (compliance). Continuous increases of volume leads to progressively larger increases of intracranial pressure. Even though the increase of volume is the same in A and B, the lower brain compliance results in higher increases of pressure in B. The curve will be steeper when the insult is acute, due to a lesser capacity of compensation.
In ALF, intracranial hypertension is usually the manifestation of an increase in water content of brain tissue.9 An increase of cerebral blood flow (CBF) is also a feature seen in many patients and in experimental models of ALF or hyperammonemia, and can contribute to the increase of intracranial pressure via an increase in cerebral blood volume. Cerebral imaging has shown a reduction of cerebrospinal fluid in ALF.9
Neuropathology of brain edema in acute liver failureAstrocyte swelling is a common finding in neuropathological studies of brain autopsy material from patients with ALF10 and from animal models of brain edema due to ALF.11-15 Swelling is more prominent in foot processes than in cell bodies, and affects predominantly astrocytes located in gray matter.11,12 In accordance with this observation, the increase in brain water content detected by the gravimetric technique in different animal models of ALF was also selective of cortical gray matter, and was absent from subcortical, mesencephalic or pontine white matter or in the cerebellum.11,12 This consistent alteration of astrocytes is in contrast with the ultrastructural preservation of neurons of the same brains.10-14
Tight junctions that characterize endothelial cells of brain capillaries were intact in all ultrastructural studies, excluding a gross disruption of the blood-brain barrier in ALF.10,11,14 However, a significant vacuolization and enlargement of endothelial cells, basement membrane and extracellular space was evident in some studies,10,13 suggesting increased permeability and pinocytic vesicular transport across the bloodbrain barrier. Several functional studies have shown increased permeability to inulin,16 sucrose,16 GABA and analogues,17,18 Trypan blue15,19 or horseradish peroxidase12 in different animal models of ALF and even in the rat 24 hours after portacaval anastomosis (PCA).12 In contrast, no abnormalities in the ultrastructure or permeability of the bloodbrain barrier are found in other studies.11,20 Differences in methodology, animal species, models used or time of sampling (before/after intracranial hypertension has developed) may be some of the reasons for the discrepancies.
Pathogenesis of brain edema in ALFFor many years, several hypotheses have tried to independently explain the occurrence of brain swelling in ALF. In the last decade, however, we have witnessed a progressive convergence of previously excluding theories to a more rational context, based on solid research observations. A critical role for ammonia is now evident. First, hyperammonemia alone appears sufficient as a cause of brain edema, as brain edema is seen in hyperammonemic patients with genetic disorders of urea cycle enzymes who do not have other alterations of liver function.21 Also, exposure to ammonia results in brain edema in vivo22,23 and astrocyte swelling in vitro.24,25 Second, ammonia appears necessary, as there are no reports of brain edema in ALF with normal ammonia levels, and increased concentrations of ammonia in blood and brain are consistent findings in experimental ALF.26,27 Furthermore, Clemmesen and cols have recently shown that cerebral herniation only occurred in their study in those patients with ALF and deep encephalopathy that had plasma ammonia levels above 150 micromols/L.28
An osmotic disturbanceAn osmotic disturbance of the astrocyte as a result of ammonia is an observation that finds most support in clinical and experimental data. The main pathway for detoxification of ammonia is through formation of glutamine as the brain lacks a complete urea cycle.29 The formation of glutamine from glutamate is catalyzed by glutamine synthetase, an enzyme located mainly in astrocytes,30 with the consumption of 1 molecule of ATP. Because glutamine is a compound with osmotic properties, its accumulation within the astrocyte has been proposed as one of the mechanism leading to astrocyte swelling.
The presence of increased brain glutamine levels in animal models and patients with ALF or hyperammonemia has been confirmed in multiple studies.22,23,26,31-37 An increased brain efflux of glutamine has been observed in patients with ALF compared to cirrhotic and healthy controls, which was higher in those who subsequently died of cerebral herniation.38 The increase of brain glutamine seems to be an early event, as evidenced by the 2-fold increase seen just 24 hours after performance of portacaval anastomosis in the rat.26 Inhibition of glutamine formation results in amelioration of ammonia-induced swelling in the rat brain in vivo22,23 and in isolated astrocytes in vitro.24
Accumulation of glutamine alone, however, does not provide a complete explanation for the development of brain edema. Mild hypothermia32 and indomethacin administration,39 for example, reduce brain swelling and intracranial hypertension in ammonia-infused PCA rats, despite similar increases of glutamine in brain. Other elements must be present to account for the development of brain swelling. One possibility would be the involvement of other organic osmoles, such as alanine. Alanine, which can be generated from transamination of glutamine, is also increased in the rat brain of ALF models.26,40 Notably, whereas glutamine increases rapidly in the early stages of HE and remains elevated to the same extent at coma stages, alanine continues to progressively rise in parallel with worsening encephalopathy.40 Other organic osmoles, such as myo-inositol or taurine, seem to be unchanged or slightly decreased in experimental models of ALF.31,41-43
The role of the blood-brain barrierAn alteration of the blood-brain barrier could explain a vasogenic theory for brain edema in ALF. Notably, functional abnormalities of the blood-brain barrier have been described in various experimental models.12,15-19 However, if an alteration of the blood-brain barrier permeability were the initial and critical event, it could hardly explain the selective swelling of astrocytes and the alterations in aminoacids and organic osmoles described above. Furthermore, no beneficial effects of corticosteroid therapy, a measure supposed to improve blood-brain barrier permeability, were seen in patients with ALF and intracranial hypertension.44 Alterations of blood-brain barrier permeability, if present, appear to play more a secondary and/or facilitating role, than being the central determinant of brain water accumulation in ALF.
Alterations of brain glutamateGlutamate is the main excitatory neurotransmitter of the brain, participating in more than 80% of synapses. Total brain glutamate levels were found to be decreased in ALF26,36,37 and in hyperammonemia.22,31,32 However, levels of extracellular brain glutamate are increased in patients45,46 and experimental models41,47-50,6-49 of ALF, as measured via brain microdialysis (a technique that allows monitoring of extracellular space composition). The increase of extracellular glutamate probably results from the impairment of glutamate re-uptake by astrocytes, given that decreased expression of astrocytic glutamate transporters as well as decreased astrocytic glutamate re-uptake have been shown in various experimental models.51-56 An increased release of glutamate is also possible.57,58
A role for glutamate in the pathogenesis of brain edema in ALF is suggested by 1) glutamate induces astrocytes swelling when injected into the brain or in isolated preparations59-61 2) brain extracellular glutamate correlates positively with severity of HE and brain water content in some ALF models,41,48 and 3) administration of glutamate antagonists has been reported to increase survival62 and to ameliorate brain edema63 in rodents with acute ammonia intoxication. Despite these observations, the exact role of glutamate in the production of brain edema in ALF requires further investigation.
Oxidative and nitrosative stressA potential role for oxidative and nitrosative stress in the pathogenesis of brain edema in ALF is being increasingly explored. In the clinical setting, treatment with the antioxidant N-acetylcysteine was associated with less frequent progression to coma and brain edema in patients with ALF.61,62 These effects, however, were thought to arise from an improved brain microcirculation.64 In our lab, treatment with N-acetylcysteine ameliorated ammonia- induced brain edema in PCA rats, despite the lack of differences in CBF or haemodynamics compared to placebo- treated controls.65 Ammonia, which has been reported to increase the formation of free radicals both in vivo66,67 and in vitro,68 could be one potential source for oxidative stress. An ammonia-induced increase of hemeoxygenase-1 gene expression, which is considered the best gene-marker of oxidative stress, also supports this assumption.69,70 Regarding nitrosative stress, increased expression and activity of neuronal nitric oxide synthase71 and increased brain nitric oxide production72 have been shown in experimental models of hyperammonemia. Evidence for nitrosative stress arises also from cellular studies, where exposure of isolated astrocytes to ammonia resulted in increased nitration of protein tyrosine residues.73 Astroglial protein tyrosine nitration was also found in brains from rats after acute ammonia intoxication or after portacaval anastomosis, indicating the possible in vivo relevance of those findings.
Even though of considerable interest, further in vitro and in vivo work is needed in order to clarify the role of oxidative and nitrosative stress in the pathogenesis of brain edema in ALF.
Energy failureMalfunction of the Na+, K+-ATPase pump by a substance not cleared by the failing liver, which would lead to accumulation of intracellular sodium, was initially suspected.74,75 This was not confirmed in subsequent studies. Depletion of brain ATP could not be demonstrated with preserved concentrations of high energy phosphates (phosphocreatine, ATP) in various experimental models of ALF.27,76,77 Thus, brain energy failure is considered to be an improbable pathogenic event, at least prior to the development of intracranial hypertension.
Cerebral blood flow and the pathogenesis of brain edema and intracranial hypertension in ALFIn patients with ALF, a wide spectrum of values of CBF has been reported, ranging from abnormally low to abnormally high levels.64,78,79 Even though differences in methodology could explain some of the discrepancies, intra- individual variations have been noted. Thus, the wide spectrum of CBF in ALF is more likely to reflect a real situation where CBF is subjected to the influence of multiple factors,80 such as disease severity, systemic haemodynamics or extrahepatic complications. Despite these variations, it is now well accepted that cerebral oxidative metabolism is preserved in ALF,81,82 with CBF usually higher than the metabolic needs of the brain (the so-called luxury perfusion).79,82
In the following paragraphs, we will examine how the normal coupling between CBF and brain metabolism is altered in ALF, and how changes of CBF can influence the development of intracranial hypertension and brain edema in ALF.
Loss of CBF autoregulation in ALFIn normal conditions, CBF varies according to the metabolic requirements of the brain,83 increasing or decreasing in parallel with brain activity. This autoregulation occurs independently of changes in mean arterial pressure or cardiac output, whenever blood pressure varies within the limits of 60 to 160 mmHg. Landmark studies by Larsen and cols have clearly shown that CBF autoregulation is lost in patients with ALF.84,85 The loss of autoregulation can be explained by the presence of vasodilatation of cerebral arterioles.86 Maneuvers that induce vasoconstriction of cerebral vessels, such as hyperventilation leading to hypocapnia, can restore CBF autoregulation in ALF.87 Restoration of autoregulation can also be achieved by moderate hypothermia88 and liver transplantation.85
Influence of CBF on brain edema in ALFIn patients with ALF, cerebral hyperemia has been associated with deeper coma, increased brain edema and higher mortality.64,79,89 Furthermore, interventions that abrogate an increase in CBF, such as hypothermia or indomethacin administration, have been shown effective for controlling or delaying the development of brain edema and intracranial hypertension in humans88,90,91 as well as in animal models of ALF.12,32,39,48 In the PCA rat with ammonia infusion, we have shown a predictable and selective rise of CBF that occur prior to the development of brain edema, in the setting of normal systemic haemodynamics.72 Three more observations in this model suggest an important role for a high CBF in the pathogenesis of brain edema: 1) there is a striking positive correlation between CBF and brain water content, 2) abrogation of the increase in CBF by indomethacin or hypothermia results in amelioration of brain edema,32,39 and 3) the increase in CBF does not occur when brain edema is prevented via the inhibition of glutamine formation with methioninesulfoximine.72 Why CBF increases in this model has not been elucidated, and even though a role of nitric oxide was initially suggested,72 subsequent studies did not confirm this contention.92
The mechanism by which CBF can influence brain edema deserves further attention. The flux of water/solutes across the blood-brain barrier is determined by the terms of the Starling equation, as recently discussed:93
Water flux = Lp x A x [ΔP - Σσ (i) x Δπi]
where Lp is the filtration coefficient of the capillary membrane, A is the capillary membrane area, ΔP is the hydrostatic pressure gradient between the lumen of the capillary and the interstitial tissue, σ (i) is the reflection coefficient of substance i over the capillary membrane, and Δπi, is the osmotic pressure gradient created by the concentration gradient of substance i between blood and tissue.
Changes in CBF and cerebral autoregulation in ALF could favour water flux into the brain by affecting mainly several components of the Starling equation. First, a higher hydrostatic pressure gradient (ΔP) is a normal result of an increase in CBF if no new capillaries are opened. Also, given that autoregulation is lost in ALF as a consequence of cerebral arteriolar vasodilatation,86 variations in systemic arterial pressure will greatly influence the hydrostatic pressure in the capillaries. Second, an increase in CBF would result in increased ammonia delivery to the brain. This could worsen Δπi by inducing further accumulation of intracellular glutamine and increase of brain tissue osmolarity, which would favour the flux of water into the brain. Finally, an increase in CBF could lead to a larger capillary membrane area (A) if new capillaries were opened (capillary recruitment), but the relevance of such mechanism in the brain is controversial.94
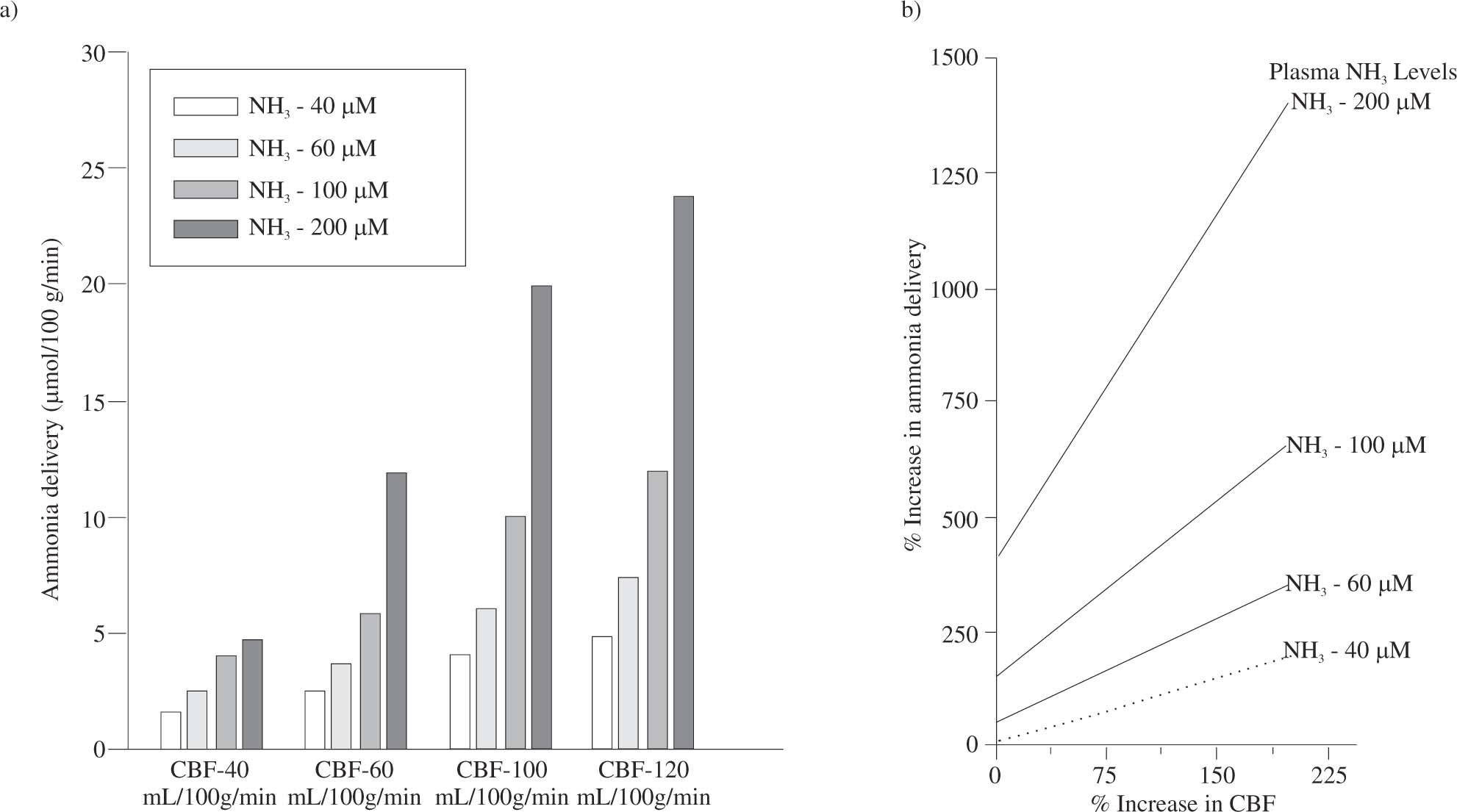
Subtle alterations in the permeability of blood-brain barrier, that would affect Lp and σ (i), have been previously discussed. The net consequence of an increase in blood flow is to raise ammonia delivery to the brain. Indeed, small increases in blood flow will greatly increase ammonia delivery to the brain in a hyperammonemic state (Figure 3).
. With increased ammonia levels, an increase in cerebral blood flow leads to disproportionate increases of ammonia delivery to the brain.")
Influence of cerebral blood flow on ammonia delivery to the brain. The right panel shows calculated absolute values of ammonia delivery for different values of cerebral blood flow and levels of plasma ammonia. In the left panel, it is shown the percentage increase of ammonia delivery relative to the normal ammonia delivery to the brain (calculated for normal ammonia plasma levels of 40 microM and cerebral blood flow of 40 mL/100 g/ min). With increased ammonia levels, an increase in cerebral blood flow leads to disproportionate increases of ammonia delivery to the brain.
All clinicians recognize that multiple factors can potentially influence the development of HE. Thus, it is not surprising that a wide array of factors can affect the expression of brain edema and intracranial hypertension in the clinical setting.92 We will examine several physiological variables, applying concepts previously reviewed.
InfectionInfection is a well-known precipitant of hepatic encephalopathy in chronic liver disease.95 In ALF, more than 80% of patients present evidence of infection,96 and increased levels of circulating cytokines are a consistent finding.97-99 A correlation between infection or the systemic inflammatory response syndrome (SIRS) and presence of severe HE has been reported in ALF.100 Furthermore, infection was an independent predictor of progression from mild to severe HE in a study of the US Acute Liver Failure Study Group.101 The mechanisms by which infection could precipitate or worsen HE are not well understood but are likely to be diverse, with effects on the periphery (deterioration of liver function, alteration of haemodynamics) and effects on the brain. Receptors for some cytokines such as IL-1 have been described in brain capillaries, suggesting that cytokines could exerts their effects in the brain even without crossing the blood-brain barrier.102
Necrotic liverImprovement of brain edema and control of intracranial pressure after total hepatectomy has been reported in patients with ALF.103,104 An increased release of cytokines from the necrotic liver suggests an inflammatory element in the intracranial hypertension of ALF.105 However, confounding factors were present in these studies, such as the use of extracorporeal detoxification methods or mild hypothermia, clouding the assessment of the effect of hepactectomy per se.
TemperatureTemperature is a component of the systemic inflammatory response syndrome. Whereas hypothermia has been shown to exert beneficial effects in decreasing brain edema and intracranial pressure in patients90 and animal models,32,48 of ALF, hyperthermia is a deleterious event. Hyperthermia was shown to precede surges of intracranial pressure in patients with ALF,106,107 to increase cerebral blood flow in dogs,108 and to increase blood-brain barrier permeability in some animal models.109 Fever should be vigorously treated in patients with ALF.
AgitationEpisodes of agitation often precede surges of intracranial pressure in patients with ALF,106 making the prevention and treatment of agitation an important aspect in the management of these patients. Agitation may reflect increase extra-cellular brain glutamate levels.48,49 Propofol is the preferred approach to handle this situation.
Arterial pressure and cerebral blood flowChanges in arterial pressure will influence CBF and intracranial pressure in the patient with ALF that has a loss of cerebral autoregulation and poor brain compliance. In this setting, even small increases of arterial pressure may lead to increased cerebral blood volume and intracranial hypertension. On the other hand, a decrease of arterial pressure in the patient with intracranial hypertension could lead to brain hypoxia. Thus, monitoring of intracranial pressure and of jugular bulb oxygen saturation has been proposed to tailor the treatment with vasoactive drugs in ALF patients.92 Cerebral perfusion pressure (mean arterial pressure minus intracranial pressure) must be maintained above 40 mmHg to avoid tissue hypoxia. Jugular bulb oxygen saturations lower than 55% are indicative of cerebral ischemia, while saturations greater than 85% indicate decreased metabolic demands of the brain or, more commonly, cerebral hyperemia.
Glucose and lactateEven though cerebral energy metabolism seems to be preserved until the last stages of the disease, brain metabolism of glucose is altered in ALF. In a preliminary report, hyperglycemia above 12 mM (200 mg/dL) was shown to be associated with intracranial hypertension in patients with ALF.110 Brain edema from other etiologies is known to worsen if hyperglycemia is present.111,112 However, mild hyperglycemia was not associated with worsening of brain edema in the ischemic rat model of ALF.113 Thus, the consequences of hyperglycemia in ALF are still unclear.
Lactate is increased in the plasma of patients with ALF114 and was initially interpreted to be the result of tissue hypoxia. Subsequent studies showed increased lactate production in the brain64 and led the authors to postulate a pathological supply dependency of oxygen to the brain.115 Increased production of lactate by the brain has been further confirmed in various experimental models of ALF.27,36,41,42,76 However, cerebral oxidative metabolism is preserved in patients with ALF.81 Two recent reports provide important insight and cast doubt on the “hypoxic” mechanistic explanation. First, Tofteng and cols. have shown an increase in extracellular lactate in patients with ALF and deep HE by means of brain microdialysis.45,46 Notably, increases in extracellular glucose and lactate preceded surges of intracranial pressure without a rise in extracellular glutamate.45 No change in lactate/pyruvate ratio and no evidence of tissue hypoxia were detected in most subjects. Based on these findings, the authors proposed that lactate may be implicated in the cerebral vasodilatation and surges of intracranial pressure in ALF. Second, Zwingman and cols. showed a selective increase of de novo synthesis of lactate from glucose in the brains of rats with acute ischemic liver failure, by means of proton and carbon-13 NMR spectroscopy.40 In contrast to the levels of glutamine, which increased in the precoma stage but did not subsequently rise, the increase in lactate correlated with the severity of encephalopathy. Based on these findings, the authors suggested that alterations of cellular glucose-and energy metabolism rather than the intracellular (astrocytic) accumulation of glutamine were the major cause of HE and brain edema in that model. Further studies to investigate the role of glucose and lactate in HE and brain edema are needed, and could have important repercussions in the clinical management of these patients.
Water and electrolyte abnormalitiesSodium concentration is the main factor determining plasma osmolarity under normal conditions. Hyponatremia is common in cirrhosis and water/electrolyte abnormalities are a common precipitant of HE. The relatively high frequency of central pontyne myelinolysis in hyponatremic cirrhotic subjects after liver transplantation116 is probably a reflection of the profound alteration of osmoregulation and osmo-compensation in the brain of these patients.117 Extreme care should be taken in the correction of hyponatremia before, during and after liver transplantation.
The consequences of hyponatremia were studied in experimental models. In the rat with portacaval anastomosis, induction of chronic hyponatremia by 1-desamino-8-Darginine vasopresin treatment decreased the levels of brain organic osmolytes.118 Infusion of ammonia resulted in the expected rise of glutamine and a reduction of taurine and myo-inositol, the main organic osmolytes in both normonatremic and hyponatremic rats. Importantly, infusion of ammonia resulted in a higher degree of brain edema in hyponatremic compared to normonatremic rats, despite an attenuated rise of glutamine in the former. These observations suggest that chronic hyponatremia makes the brain more vulnerable to an ammonia-induced osmotic disturbance with subsequent brain swelling. In contrast, increasing plasma osmolarity by infusion of hypertonic saline has been observed to reduce intracranial hypertension in ALF,93 its use is being evaluated in ongoing clinical trials.
Alterations of potassium could also influence HE by altering normal ammonia metabolism. Hypokalemia increases renal ammoniagenesis and seems to result in both increased urinary excretion and venous secretion of ammonia as seen in humans with chronic potassium depletion.119 Serum potassium and urine flow significantly correlated with kidney ammonia production, with serum potassium accounting for 61.4% of variations in ammonia production.
Ammonia metabolismA highly orchestrated interorgan metabolism and trafficking of ammonia and glutamine is present in physiological conditions. Chronic and acute liver failure result in hyperammonemia and leads to an important disturbance of normal body nitrogen homeostasis (for review see).120 Different clinical factors influence per se the plasma levels or the effects of ammonia in the brain in liver failure.
Muscle wastingEven though glutamine synthetase activity in skeletal muscle is low, its relative increased mass compared to other organs that contain this enzyme makes the muscle one of the main glutamine synthesizing organs. Due to the increased plasma ammonia levels in liver failure, the muscle becomes an important ammonia detoxifying organ.121 Avoidance of muscle wasting in cirrhosis and stimulation of ammonia metabolism in the muscle are important potential therapeutic targets in chronic and acute liver failure.
Gastrointestinal bleedingGastrointestinal bleeding is a known precipitant of HE. Blood in the digestive tract has been shown to be a potent stimulus for the intestinal production of ammonia.122 In the rat with portacaval anastomosis, simulated gastrointestinal bleeding led to increased levels of ammonia and glutamine in the brain and to worsening of hepatic encephalopathy,123 indicating a mechanistic association.
Acid-base abnormalitiesAmmonia can occur in blood as ammonium ion (NH4+) or as ammonia gas (NH3). The ammonia gas form is lipophilic and diffuses easily across cell membranes, whereas the ammonium ion is non-diffusible and crosses membranes only by carrier-mediated processes.124 At physiological pH, most ammonia is in the ion form and only 1% is present as ammonia gas. Circumstances that alter blood pH could, thus, be pathogenically important. Metabolic or respiratory alkalosis, produced for example by diuretics or hyperventilation, could increase the gaseous form of ammonia, facilitating in this way its entry into the brain. However, the global effect of an altered pH in ammonia metabolism is difficult to evaluate, because the consequences can be different in different organs. Thus, whereas alkalosis can favour ammonia detoxification by improving urea synthesis in the liver,125 it may increase ammonia production in the kidney.126,127 Similarly, whereas metabolic acidosis increases gut release of ammonia, it increases ammonia uptake in the liver and the muscle.128,129
SedativesThe role of sedatives as precipitants of HE is well recognized. Recent reports implicate endogenous benzodiazepine- like compounds in the pathogenesis of HE and brain edema. In isolated astrocytes, different benzodiazepines induced protein tyrosine nitration130 and free radical production131 in a manner similar to that produced by ammonia68,73 and exacerbated ammonia-induced astrocyte swelling.132 Sedation is difficult to avoid in the agitated patient with ALF, but benzodiazepines should be avoided.
ConclusionHepatic encephalopathy of chronic liver disease and brain edema of acute liver failure have been considered two distinct and unrelated clinical entities for many years. Even though their pathogenesis is still not fully resolved, a new paradigm has progressively developed where both entities are different elements of a continuous spectrum. Hepatic encephalopathy and brain edema seem to share common pathogenic roots, with a key role of ammonia and a critical involvement of the astrocyte in both complications. The study of one helps to understand the other: whereas ALF facilitates the study of causal relationships in a firmer manner, chronic liver failure gives the opportunity to study brain compensating mechanisms. This integrated view provides also a better perspective to judge the pathophysiological relevance that other factors may have in the manifestation of the disease.



