



Polycystic liver disease (PLD) is a celiopathy characterized by progressive growth of multiple hepatic cysts. In a minority of patients, severe symptomatic hepatomegaly necessitates liver transplantation (LT). The purpose of this study is to describe the postoperative and long-term outcomes of all patients transplanted for PLD at our center.
All patients who underwent LT for PLD were identified through our database. Using patient charts, data were extracted on patient demographics and medical history, postoperative surgical and medical complications, length of hospitalization, prevalence of chronic kidney failure, and patient and graft survival. Subjects were contacted in April 2010 to verify their survival and confirm their need, if any, for hemodialysis and/or kidney transplantation. Descriptive statistics for patient and graft survival were performed. From 1993 to 2010, 14 subjects underwent LT and 1 subject underwent combined kidney and LT; all subjects were female and the mean age was 49.0 years. 10 (66.7%) subjects had polycystic kidney disease. Patients experienced a high rate of vascular complications, including hepatic artery thrombosis (HAT) or stenosis in 3 (20%) and 2 (13.3%) subjects, respectively. One subject had early graft loss due to HAT and underwent re-transplantation. The mean length of hospitalization was 18.8 days. After a mean of 66.8 months of follow-up (3-200), 13 (86.7%) subjects are alive with satisfactory graft function, and no patients had renal failure.
In conclusion, patients who underwent LT for PLD had a high rate of postoperative vascular complications. However, long-term patient and graft survival, and kidney function, is excellent.
Polycystic liver disease (PLD) is a celiopathy caused by defects in polycystin proteins. The autosomal dominant (AD) form of the disease is characterized by the development of multiple simple cysts in the liver that progress in size over time. Although an autosomal recessive (AR) form of PLD exists, this condition is rare and is associated with autosomal recessive polycystic kidney disease (ARPCKD), congenital hepatic fibrosis, intrahepatic biliary dilatation and a high mortality in infancy. As such, in adulthood only AD PLD is seen. Autopsy series suggest that the prevalence of PLD is 0.13%.1
When PLD occurs in association with autosomal dominant polycystic kidney disease (ADPCKD), the pathogenesis involves defects of polycystin-1 or, less commonly, polycystin-2.2 In contrast, when AD PLD has no concurrent renal involvement, mutations of the gene protein kinase substrate 80K-H (PR-KCSH), which encodes the protein hepatocystin, are implicated.3 Polycystin proteins are integral to the function of cilia lining cholangiocytes. In the setting of defective cystoproteins in cilia, cyst formation occurs through a variety of mechanisms, including enhanced cell proliferation and apoptosis; increased secretion of fluid; and faulty cell adhesion and ciliary function.1 Estrogens and a variety of growth factors also play a role in cyst proliferation and secretion.2
PLD has a broad spectrum of clinical manifestations. Clinically, PLD is defined as 5 or more simple cysts greater than I cm in diameter, in the absence of infection or trauma. Interestingly, patients with more severe liver involvement from PLD may be predisposed to more significant renal dysfunction from ADPCKD.4 Conversely, patients with more extensive renal cystic disease develop more liver cysts.5 PLD is more severe in women, likely because of the hypothesized role played by female sex hormones in the pathogenesis of liver cyst formation.
While liver failure is uncommon from PLD, and most patients require no radiologic or surgical intervention, in a minority of patients liver cysts cause significant morbidity.6 Compressive symptoms produced by hepatic cysts can result in gastrointestinal obstruction, hepatic venous outflow obstruction, and compression of the inferior vena cava, portal vein and bile ducts. Other cyst complications include hemorrhage, infection, torsion or rupture.
Although there are no proven medical treatments for PLD, surgical techniques involving hepatic volume reduction are the cornerstone of care for the rare patient with severe symptoms due to innumerable large cysts unsuitable for cyst aspiration and sclerosis. Operative techniques include partial hepa-tectomy with remnant cyst fenestration, cyst fenes-tration alone, or LT. While there are no strict criteria for LT for PLD, generally transplantation is reserved for patients with significant morbidity from severe cystic disease refractory to other radiologic and surgical techniques. Such patients typically have no intrinsic liver dysfunction but have a poor quality of life due to symptoms such as dyspnea, early satiety, and abdominal and back pain. Many liver transplantation allocation systems (e.g. United Network for Organ Sharing; Trillium Gift of Life Network Ontario) provide exception points for listing patients with PLD following careful consideration by a multidisciplinary committee on a case-by-case basis.
As PLD is an uncommon albeit established indication for LT, there is limited information on immediate post-operative as well as long-term clinical outcomes. In a German series of 36 subjects, Kirch-ner et al reported that recipients transplanted for PLD reported an improved quality of life and had excellent long-term survival, although 5 (13.9%) subjects died of medical complications within 2 months of transplantation.7 Similarly, in amongst largest series published in North America to date of 13 subjects transplanted for PLD, Taner, et αl., reported 3 deaths in the postoperative period, and 9 post-transplant surgical procedures for 5 patients due to surgical complications (bowel perforation, bile leak).8 If patients survived the post-surgical period, however, excellent long-term patient and graft survivals were seen.8
One may postulate that patients with PLD may have a complex operative course due to the challenges in resecting a massive native liver with distortion of vascular structures. Moreover, given the strong correlation of ADPCKD, it is conceivable that patients transplanted for PLD may experience higher rates of renal failure over time. Further data are needed to explore these hypotheses and corroborate with the findings of prior studies.
AimThe purpose of this study is to describe the postoperative and long-term clinical outcomes of all patients transplanted for PLD at a Canadian transplant center.
MethodsA retrospective cohort study was conducted on all adult patients transplanted for PLD from January 1993 to February 2010 at London Health Sciences Center, London, Canada. All subjects provided consent, and approval was obtained by the institutional ethics committee. Patients were identified through the institutional liver transplant database. Using hospital health records, information was gathered on patient demographics (date of birth, race, sex, medical comorbidities), baseline liver and kidney function, date of LT, type of LT (deceased, deceased after cardiac death, or live donor), size and length of explanted liver, postoperative surgical and medical complications, re-operation (including re-transplantation), length of hospitali-zation, and re-admission within 3 months. Patient charts were also examined to document long-term medical complications, namely new onset renal failure (defined as need for hemodialysis or kidney transplantation). All subjected were contacted in April 2010 to verify their survival and their need, if any, for long-term hemodialysis or kidney transplantation. Descriptive statistics were reported as frequencies, means and ranges. Statistical analysis was performed using STATA 11.0.
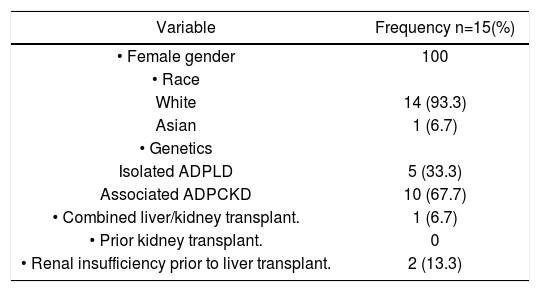
ResultsFrom January 1993 to February 2010, 14 patients underwent LT and 1 patient underwent combined kidney and LT for polycystic disease. Baseline characteristics are summarized in table 1. All subjects were female, with mean age 49.0 (40.0-65.0) at the time of LT. No subjects underwent prior cyst fenestration and/or partial hepatectomy. All recipients received deceased grafts. 14 (93.37) subjects were Caucasian and 1 (6.7%) subject was Asian. 10 (66.7%) patients had concomitant ADPCKD. The mean creatinine just prior to LT was 112.9 umol/L (43-209).
Baseline characteristics.
| Variable | Frequency n=15(%) |
|---|---|
| • Female gender | 100 |
| • Race | |
| White | 14 (93.3) |
| Asian | 1 (6.7) |
| • Genetics | |
| Isolated ADPLD | 5 (33.3) |
| Associated ADPCKD | 10 (67.7) |
| • Combined liver/kidney transplant. | 1 (6.7) |
| • Prior kidney transplant. | 0 |
| • Renal insufficiency prior to liver transplant. | 2 (13.3) |
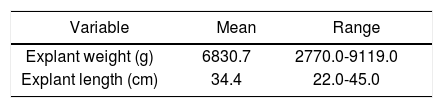
The weight and length of the native liver following resection are summarized in table 2. The mean weight of the explanted liver was 6830.7 g (2770.09119.0). The mean diameter of the explanted liver was 34.4 cm (22.0-45.0).
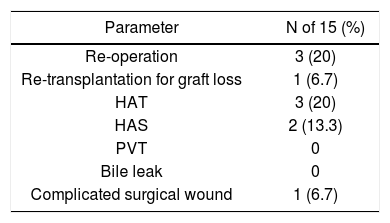
Postoperative complications are shown in table 3. Overall, 5 of 15 subjects had vascular complications, including 3 (20%) and 2 (13.3%) subjects with hepatic artery thrombosis (HAT) and hepatic artery stenosis (HAS), respectively. No patients had portal vein thombosis (PVT) or bile leak. One (6.7%) patient had significant wound complications. Overall, 3 (20%) patients needed to return to the operating theatre for management of HAT. One subject required re-transplantation due to early graft loss from HAT at day 3 post-transplantation. The mean length of hospitalization was 18.8 (7-49) days. Postoperative medical complications were not observed at an increased frequency, and no patients were re-admitted to the hospital within 3 months of discharge.
All patients received long-term follow-up care. The mean follow-up was 66.7 months (3-200). The I and 5-year patient survival was 100%. The I and 5-year graft survival was 93.3%. One subject died 8 years after LT at the age of 57 due to trauma; and a second subject died 7 years after LT at the age of 67 due to natural causes, though no autopsy report was available. Thirteen subjects were contacted directly in April 2010 and their survival was verified. No patient, including those who have deceased, developed renal failure necessitating hemodialysis or kidney transplantation, or chronic hepatic allograft dysfunction warranting liver re-transplantation.
DiscussionIn this series of 15 subjects transplanted for PLD, 1 patient experienced early graft loss due to HAT, but all subjects had excellent long-term survival. The 1-year and 5-year patient and graft survival was 100% and 93.3%, and 100% and 93.3%, respectively. This compares favorably to our center’s overall 1-year patient and graft survival of 91% and 88%, and 5-year patient and graft survival of 86% and 81%, respectively, from 1999 to 2008 (n = 619, unpublished data).
However, subjects experienced a higher than expected rate of HAT at 20% and HAS at 13.3%; this compares unfavorably to our center’s overall rate of HAT of 5% from 1999-2008 (unpublished data). Given the retrospective nature of this study, a detailed analysis into the possible etiology of HAT was not feasible. Nevertheless, we postulate that because patients with PLD do not have an intrinsic coagulopa-thy like patients with hepatic dysfunction, perhaps the clamping of the hepatic artery during the transplantation may predispose to thrombosis. Furthermore, given the excess space in the abdominal cavity following resection of the native liver, there is a theoretical risk of torsion or volvulus of the hepatic graft, compromising hepatic artery blood flow. To minimize operative complications, in patients with extensive cysts incorporating the cava, caval interposition with veno-venous bypass may be preferred to the piggyback technique.
Post-operative medical complications were not observed at an increased frequency. Subjects had a mean of 18.8 days in hospital. This length of hospi-talization is significantly longer than our mean length of stay of 8.5 days following an uncomplicated LT from 1999 to 2008 (unpublished data). The longer hospitalization was observed in subjects with postoperative vascular complications requiring surgical repair.
None of the subjects had significant long-term renal impairment requiring hemodialysis or kidney transplantation, despite the fact that 2-thirds of the subjects had concomitant ADPCKD. We hypothesize that a possible reason for low rates of severe renal injury is our common practice to continue mycofeno-late mofitil and/or low dose prednisone long-term, and minimize calcineurine inhibitors in recipients at higher risk for renal injury.
A strength of this study is that we report on a well-sized cohort of subjects who underwent LT for a relatively rare indication. Furthermore, all subjects received long-term follow up, thereby enabling accuracy in the reporting of clinical outcomes such as patient and graft survival, and the development of severe renal disease. A limitation of the study is that that the small sample size precludes multivaria-te analysis.
A multicenter study to examine postoperative clinical outcomes following LT for PLD is needed to verify the findings of this study, especially the higher rate of HAT. In conclusion, despite increased rates of post-operative vascular complications in patients transplanted for PLD, high rates of patient and graft survival are seen. Furthermore, renal failure is not observed at an increased frequency, even among patients with co-existent PCKD.
Abbreviations- •
AD: Autosomal dominant.
- •
ADPCKD: Autosomal dominant polycystic kidney disease.
- •
CI: Calcineurin inhibitor.
- •
DDLT: Deceased donor liver transplantation.
- •
HAS: Hepatic artery stenosis.
- •
HAT: Hepatic artery thrombosis.
- •
LDLT: Live donor liver transplantation.
- •
LT: Liver transplantation.
- •
PCKD: Polycystic kidney disease.
- •
PLD: Polycystic liver disease.
- •
PVT: Portal vein thrombosis.
None for all authors.
AcknowledgmentThe authors thank Dr. Mamoun Al-Basheer, Dr. William Wall, Mr. Paul Myers and Mr. Michael Blo-ch for their assistance in the preparation of the manuscript.



