





Over the past decades, many drugs have been identified, that can potentially induce steatohepatitis in the predisposed individual. Classically this has been incriminated to amiodarone, perhexiline, and 4,4’-diethylaminoethoxyhexestrol (DH), all of which have been found to independently induce the histologic picture of non-alcoholic steatohepatitis (NASH). Pathogenetic mechanisms of hepatotoxicity although still evolving, demonstrate that mitochondrial dysfunction, deranged ATP production and fatty acid catabolism likely play an important role. Drugs like steroid hormones can exacerbate the pathogenetic mechanisms that lead to NASH, and other drugs like tamoxifen, cisplatin and irenotecan have been shown to precipitate latent fatty liver as well. Further research aiming to elucidate the pathogenesis of drug-induced steatosis and steatohepatitis is needed in order to better design therapeutic targets.
Non-alcoholic fatty liver disease (NAFLD) is one of the most common chronic liver diseases of western world. It is defined as fat deposition of hepatocytes exceeding 5% of the hepatocytes histologically in individuals with little or no alcohol consumption. It is also the most prevalent cause of chronic elevation of liver enzymes in the adult population as evident in National Health and Nutrition Examination Surveys (NHANES) conducted between 1988 and 2008.1 Hepatic steatosis is at the lower end of the spectrum of NAFLD clinical severity. Predominantly large (macro-) and occasionally small (micro-) vesicles of fat, mostly triglycerides, accumulate with- in hepatocytes without causing appreciable hepatic inflammation, liver cell death, or scarring. At the midrange of the severity spectrum is steatohepatitis (nonalcoholic steatohepatitis, or NASH), which is an intermediate form of liver damage superimposed upon a background of hepatic steatosis. Steatohepatitis is characterized by the appearance of focal hepatic inflammation and hepatocyte apoptosis and death. At the highest end of the severity spectrum is cirrhosis. By the time this degree of architectural distortion develops, hepatic steatosis has often disappeared. Because all of these histologic features also occur in alcohol- or drug-induced fatty liver diseases, liver biopsy cannot reliably distinguish among the various causes of this entity.
NAFLD was traditionally categorized as primary or secondary depending on the underlying etiology. Primary NAFLD is closely associated with insulinresistance and metabolic syndrome. Currently the term secondary NAFLD is discouraged and the preferred nomenclature includes the known causative factor and the resultant pathology, e.g. total parenteral nutrition-induced, drug-induced steatosis/steatohepatitis.2 Several studies have reported that liver steatosis is a common histological feature of chronic hepatitis C infection. In 2014, Abenavoli, et al. examined the relationship between hepatitis C virus and insulin resistance, looking into the role of genotype 3 in the development of hepatic steatosis.
The diagnosis of NASH is established by the presence of a characteristic pattern of steatosis, inflammation and hepatocellular ballooning on liver biopsies in the absence of significant alcohol consumption. The most commonly used tool for histologic evaluation is the NAFLD Activity Score (NAS).4 The NAS, defined as the un-weighted sum of the scores for steatosis (0-3), lobular inflammation (0-3), and ballooning (0-2), thus ranging from 0 to 8. The score was developed as a tool to measure changes in NAFLD during trials. Some studies have used threshold values of the NAS to define the severity, specifically NAS ≥ 5 as surrogate for the histologic diagnosis of NASH, however biopsies with lower scores (≤ 3) were generally not considered to be diagnostic of steatohepatitis.5
The focus of this review is to educate readers about different classic drugs known to cause steatosis/steatohepatitis in absence of alcohol intake, and review the pathogenesis and management of drug-induced fatty liver disease.
DefinitionDrug-induced fatty liver disease is a relatively rare entity identified when steatohepatitis appears to result from a direct toxic effect of a drug on the liver. Most commonly it is associated with prolonged intake of the offending medication. Approximately 2% of fatty liver disease cases are estimated to be drug-induced, confirming that a fatty liver is a rare manifestation of drug toxicity.6
Drug-induced fatty liver is strongly associated with duration and dose of medication. Some drugs induce an acute energy crisis by interrupting adenosine triphosphate (ATP) synthesis by mitochondria, resulting in microvesicular steatosis.7 Drugs can cause both micro- and macro-vesicular steatosis but usually begins acutely with microvesicular lesions (Table 1).
Drugs causing microvesicular steatosis, macrovesicular steatosis, phospholipidosis, steatohepatitis and obesity/insulin resistance.
| Drug | Macrovesicular steatosis | Microvesicular steatos | Phospholipidosis | Steatohepatitis | Obesity, insulin resistance |
|---|---|---|---|---|---|
| Amiodarone | * | * | * | * | |
| Aspirin | * | ||||
| Ibuprofen | * | ||||
| AZT | * | ||||
| NRTI (ddI, d4T) | * | * | * | ||
| Protease inhibitors | * | ||||
| Valproic acid | * | * | * | ||
| Carbamazepine | * | ||||
| Perhexiline maleate | * | * | * | ||
| DH | * | * | |||
| Tamoxifen | * | * | |||
| Methotrexate | * | * | |||
| 5-FU | * | * | |||
| Irinotecan | * | * | |||
| Glucocorticoids | * | * | * |
NRTIs: nucleoside reverse transcriptase inhibitors. ddI: didanosine. d4T: stavudine. 5-FU: 5-fluorouracil. DH: diethylaminoethoxyhexestrol.
In microvesicular steatosis, hepatocytes are filled up with numerous small lipid vesicles, which leave the nucleus in the center of the cell. Microvesicular steatosis is related to severe impairment of the mitochondrial beta-oxidation of fatty acids.8 Because non-esterified fatty acids are poorly oxidized by mitochondria, they undergo increased esterification into triglycerides, the main lipid form that accumulates in these conditions. Significant necrosis, cholestasis, and fibrosis are usually absent in acute microvesicular steatosis, as the lesion progresses rapidly to either death or resolution.7 Drugs linked to microvesicular steatosis include valproic acid, tetracycline, aspirin, ibuprofen, zidovudine and vitamin A.
Macrovesicular steatosisIn contrast, macrovesicular steatosis has a single, large vacuole of fat (mainly triglycerides), which fills up the hepatocyte and displaces the nucleus to the periphery of the cell. It usually follows a more indolent course. Macrovesicular steatosis can be seen in association with nitrofurantoin, gold, methotrexate, glucocorticoids, estrogens, acetaminophen, non-steroidal anti-inflammatory drugs (NSAIDs) such as ibuprofen, indomethacin and sulindac, antihypertensives such as metoprolol, chlorinated hydrocarbons such as carbon tetrachloride and chloroform, chemotherapeutic agents such as 5-fluorouracil and cisplatin, and tamoxifen.9
PhospholipidosisThe term drug-induced phospholipidosis ascribes to a benign condition that has been defined by the appearance of intracellular accumulation of phospholipids and lamellar bodies. The appearance of microscopic subcellular structures induced by a variety of drugs led to recognition of a disorder that was subsequently termed drug-induced phospholipidosis (DIPL).10 Ultra structural investigations revealed that these cytosolic inclusions consist of concentric myelin-like structures, the so-called lamellar bodies, the presence of which became the morphological hallmark of phospholipidosis.10
So far, more than 50 novel chemical entities have been identified to induce phospholipidosis.11 These include antibiotics, antidepressants, antipsychotics, and antimalarial and antiarrhythmic drugs. Clinically relevant phospholipidosis has been observed under administration of amiodarone, fluoxetine, gentamicin, perhexiline and 4.4’-dieethylaminoethoxyhexestrol (DH).12
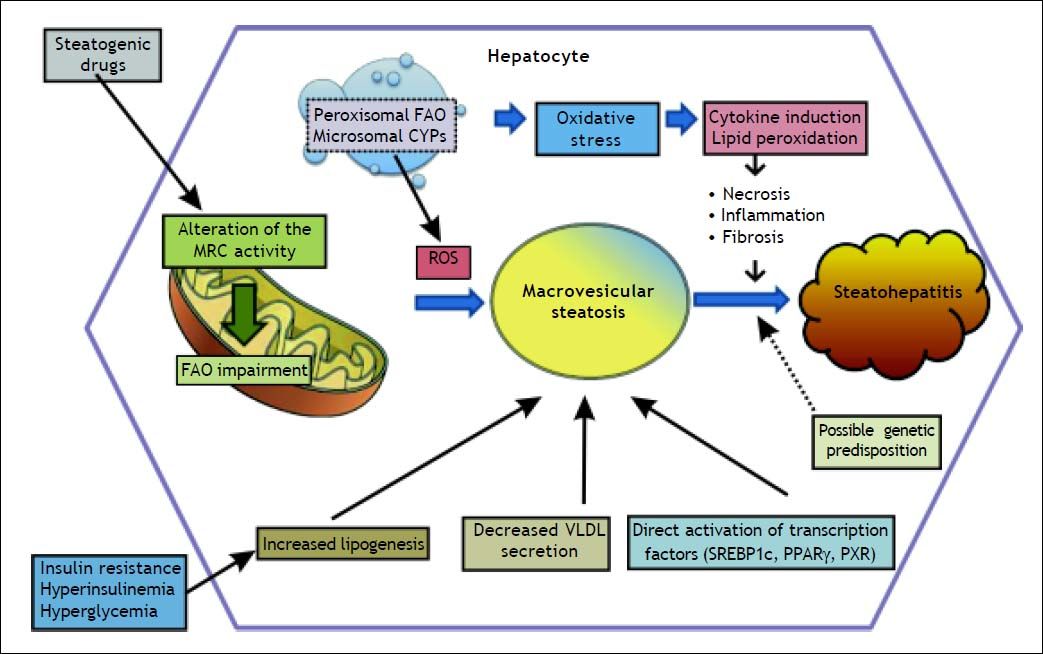
PathogenesisIn order to better understand the factors contributing to drug-induced fatty liver, a brief review of lipid metabolism will be useful. Mitochondria dysfunction is the main element associated with drug-induced fatty liver, via the direct or indirect effects of oxidative stress such as impairment of electron flow along the respiratory chain, and leakage of reactive oxygen species (ROS) produced primarily by the mitochondria (Figure 1).13
 by alteration of the mitochondrial respiratory chain (MRC) activity, reducing secretion of very-low density lipoprotein (VLDL), by directly activating transcription factors involved in hepatic lipogenesis, such as SREBP-1c, PPARγ, and PXR, and by favoring the occurrence of insulin resistance, hyperinsulinemia, and hyperglycemia. Progression from steatosis into steatohepatitis in genetically predisposed individuals involves the production of reactive oxygen species (ROS), which is responsible for oxidative stress and lipid peroxidation leading to induction of cytokines such as TNFα and TGF β, and lipid peroxidation products such as malodiealdehyde (MDA), and 4 hydroxynonenal (HNE) leading to necroinflammation and fibrosis. Production of ROS although occurs mostly through alteration of the MRC, other sources such as peroxisomal FAO and microsomal cytochromes P450 (CYPs) also contributes.")
Schematic representation of drug-induced macrovesicular steatosis and steatohepatitis. Steatogenic drugs can cause macrovesicular steatosis by impairment of mitochondrial fatty acid oxidation (FAO) by alteration of the mitochondrial respiratory chain (MRC) activity, reducing secretion of very-low density lipoprotein (VLDL), by directly activating transcription factors involved in hepatic lipogenesis, such as SREBP-1c, PPARγ, and PXR, and by favoring the occurrence of insulin resistance, hyperinsulinemia, and hyperglycemia. Progression from steatosis into steatohepatitis in genetically predisposed individuals involves the production of reactive oxygen species (ROS), which is responsible for oxidative stress and lipid peroxidation leading to induction of cytokines such as TNFα and TGF β, and lipid peroxidation products such as malodiealdehyde (MDA), and 4 hydroxynonenal (HNE) leading to necroinflammation and fibrosis. Production of ROS although occurs mostly through alteration of the MRC, other sources such as peroxisomal FAO and microsomal cytochromes P450 (CYPs) also contributes.
Under normal fed conditions, dietary triglycerides and surplus carbohydrates are converted into free fatty acids (FFA). During periods of fasting or starvation, the triglycerides stored in adipose tissue are hydrolyzed to FFAs and transported to the liver, where they are used to form phospholipids and cholesterol esters or converted into ketone bodies to be used as fuel by extra hepatic tissues.14 Potential accumulation of fat in the liver could occur when there is either increased delivery of FFA from peripheral adipocytes or the diet to the liver, increased endogenous hepatic synthesis of FFA, decreased disposal of FFA as low-density lipoprotein and very low-density lipoprotein, or a combination thereof. In contrast to this simplistic model of fatty liver disease several studies have reported an increased secretion of very low-density lipoprotein (VLDL) in subjects of fatty liver suggesting a more complex underlying pathogenetic mechanism than could be speculated.15,16 Hepatocytes play a central role in lipid metabolism within the liver. Fatty acids are oxidized by the mitochondria, peroxisomes, and microsomes. Oxidation of short-, medium-, and long-chain fatty acids occurs in mitochondria. Very long chain fatty acids (VLCFAs) are oxidized by peroxisomes. Long chain fatty acids (LCFAs) and VLCFAs are also metabolized as a result of microsomal oxidation, resulting in the production of dicarboxylic acids that are further degraded by peroxisomes. After the VLCFAs and LCFAs chains are shortened by the extra mitochondrial (peroxisomal and microsomal) oxidation, the mitochondrial oxidation system completes the oxidation process. Thus, mitochondria play a dominant role in fatty acid oxidation and are responsible for the majority of disturbances occurring in lipid metabolism.
Function of mitochondria in hepatocytesNormal hepatocyte mitochondria metabolize free fatty acids through β-oxidation, a process quantitatively much more important in the production of ATP than the hydrolysis of glucose.15 Short- (4-6 carbon) and medium- (6-14) chain fatty acids cross the mitochondrial membranes unimpeded, whereas long-chain fatty acids (14-18 carbons) require transport across the mitochondrial membrane. Transport of long-chain fatty acids involves esterification to coenzyme A in the cytosol and transport by membrane translocates into the mitochondrial matrix. After gaining access to the mitochondrial matrix, all fatty acids enter the β-oxidation cycle as CoA thioesters, or they may become esterified (stored) in triglycerides.
The β-oxidation of fatty acyl CoA esters involves four steps within mitochondria: two oxidation steps, a hydration step, and thiolysis, yielding acetyl-CoA and a fatty acyl CoA shortened by two carbons, the latter of which repeats identical cycles. Each β-oxidation cycle transfers electrons to one molecule each of NAD and FAD, yielding NADH and FADH2, respectively. Each acetyl-CoA entering the citric acid (Krebs) cycle is principally derived from glycolosis, which yields three additional molecules of NADH and one molecule of FADH2. Enzymes involved in the β-oxidation of fatty acids and in the citric acid cycle are either soluble within the mitochondrial matrix or are bound to the inner surface of the inner mitochondrial membrane.13
Subsequently, mitochondria produce ATP through the transfer of electrons from NADH and FADH2 to molecular oxygen, producing water. This oxidation of NADH and FADH2 results in a stepwise transfer of electrons through protein complexes of the respiratory chain, which exist within the inner mitochondrial membrane. During electron transport in the respiratory chain, protons are actively pumped into the intramembranous space, creating a concentration gradient and electrical potential across the inner membrane. ATP synthesis requires the entry of protons down this electrochemical gradient through the inner mitochondrial membrane and into the matrix by way of a channel of ATP synthase. These processes of electron trans- port, proton pumping, and generation of ATP are energetically coupled, and drugs that interfere with these processes are believed to uncouple respiration (dissipate the membrane proton gradient without producing ATP).
Mitochondrial dysfunction, drug-induced cytolytic hepatitis and microsteatosisThe concept that drugs can disturb mitochondrial function, leading to clinically recognizable disease entities is not new. As early as the 1970’s, a Reyelike syndrome was observed in patients treated with valproic acid for instance.17,18 The past few decades have seen, however, remarkable progress in the understanding of the different mechanisms leading to mitochondrial dysfunction, and their role in drug-induced liver disease. While membrane permeabilization, drug-induced oxidative phosphorylation (OXPHOS) impairment, and mitochondrial DNA (mtDNA) depletion have been directly linked to cytolytic hepatitis - and its full spectrum, from mild elevation in transaminases to fulminant hepatitis, drug-induced microsteatosis occurs in the setting of severe mitochondrial fatty acid oxidation (FAO) impairment. Specifically, there are four general mechanisms (Figure 1) which compromise mitochondrial FAO, eventually leading to microsteastosis.19
First, drugs can directly inhibit one or several mitochondrial FAO enzymes. This is seen with amio- darone, tamoxifen, and valproic acid (VPA). Second, drugs can indirectly impair mitochondrial FAO by impairing the generation of its major cofactors: coenzyme A and L-carnitine esters, such as seen with VPA.19 Thirdly, drugs can inhibit mitochondrial respiratory chain (MRC), impairing the regeneration of FAD and NAD +, thereby also impairing FAO; such is the case with amiodarone, perhexiline and tamoxifen.20–23 Lastly, drugs can inhibit mtDNA. Profound mtDNA depletion induces MRC impairment, leading to FAO inhibition and resultant microsteatosis.8,24 Antiviral drugs such as azidothymidine (AZT), stavudine (d4T) and didanosine (DDI) all inhibit mtDNA polymerase gamma.25–27 Other than direct inhibition of mtDNA polymerase, mtDNA levels can be indirectly inhibited by drugs, which leads to TCA cycle inhibition and then lactic acidosis,28,29 and direct mtDNA damage itself can occur from ROS, reactive nitrogen species (RNS), or drug metabolites.30
Several drugs known to cause steatosis/steatohepatitis and their corresponding effects on mitochondrial functions has been summarized in table 2.
Hepatotoxic drugs known to cause steatosis/steatohepatitis and their corresponding effects on mitochondrial function.
| Drug | MPTP opening | Direct inhibition of mitochondria FAOa | OXPHOS uncoupling | Direct inhibition of the MRC b | mtDNA depletion/damagec | References |
|---|---|---|---|---|---|---|
| Acetaminophen (APAP) | * | * | * | 156, 157 | ||
| Ibuprofen | * | * | 158, 159 | |||
| Diclofenac | * | * | * | 159, 160 | ||
| Salicylic acid | * | * | * | 159, 161, 162 | ||
| Amiodarone | * | * | * | * | 8, 21, 163 | |
| Stavudine (d4T) | * | 164 | ||||
| Zidovudine (AZT) | * | 164 | ||||
| Valproic acid | * | * | 165, 166 | |||
| Perhexiline maleate | * | * | * | 22 | ||
| DH | * | * | 115 | |||
| Tamoxifen | * | * | * | * | 19, 23 | |
| Methotrexate | * | * | 167 | |||
| 5-FU | * | 24, 168 | ||||
| Irinotecan | * | * | * | 24, 168 |
FAO: fatty acid oxidation. MPTP: mitochondrial permeability transition pores. MRC: mitochondrial respiratory chain. mtDNA: mitochondrial DNA. OXPHOS: oxidative phosphorylation. DH: diethylaminoethoxyhexestrol.
Macrovesicular steatosis (macrosteatosis), generally known as fatty liver, is commonly observed in both alcoholic and non-alcoholic liver disease. In the setting of non-alcoholic fatty liver, it is associated with cardiometabolic risk factors such as obesity and diabetes.31 Macrosteatosis can also be druginduced, and there are four known mechanisms involved. First, similar to microsteatosis, inhibition of mitochondrial FAO can also lead to macrosteatosis.20,22,24 Secondly, microsomal triglyceride transfer protein (MTP) can be directly inhibited by drugs such as amiodarone and perhexiline, leading to macrovesicular steatosis.8,32 Increased cellular uptake of fatty acids is the third mechanism. A well demonstrated example of such is seen with the drug efavirenz, which activates AMP-activated protein kinase (AMPK) leading to efavirinez-induced mitochondrial dysfunction.33,34 Finally, drugs can directly stimulate lipid synthesis in the liver; while the direct mechanism of such stimulation is unknown, the activation of lipogenic transcription factors, such as PXR, PPARγ and glucocorticoid receptor have been implicated.35–39
Drug-induced phospholipidosisAs previously mentioned, clinically relevant phospholipidosis has been associated with drugs such as amiodarone, fluoxetine, gentamicin, perhexiline and DH. Many of these are cationic amphiphilic drugs (CADs) that share particular physical properties resulting from a chemical structure containing a hydrophilic ring and hydrophobic regions. Several of these drugs display severe adverse effects, such as acute pneumonitis or hepatitis as observed under amiodarone treatment.40 CAD-induced phospholipidosis is characterized by four principal features; excessive accumulation of phospholipids in cells; ultrastructural appearance of membranous lamellar inclusions, predominantly lysosomal in origin; accumulation of the inducing drug in association with the increased phospholipids; and reversibility of alterations after discontinuance of drug treatment.41
Basically two hypotheses have been put forward to explain the underlying mechanisms of drug-induced phospholipidosis.41 The first hypothesis assumes that CADs bind directly to phospholipids to result in indigestible drug-lipid complexes, which accumulate and are stored in the form of lysosomal lamellar bodies.13 The second hypothesis is based on the observation that production of lamellar bodies was associated with inhibition of phospholipase activity; either due to direct inhibition or interaction of CAD at the phospholipid bilayer of the lysosome.41 The functional consequences of the presence of this condition on cellular or tissue function are not well understood. The general consensus is that the condition is an adaptive response rather than a toxicological manifestation; however, additional studies to examine this question are needed. Until this issue is resolved, concerns about phospholipidosis will continue to exist at regulatory agencies. Procedures for the screening of potential phospholipogenic candidate compounds based on gene expression analysis in HepG2 cells have been shown.42
Progression from Steatosis into SteatohepatitisOverviewWhile the exact mechanisms by which steatosis progresses into steatohepatitis are not clearly delineated, a key role of mitochondrial dysfunction has been proposed. Drugs that impair mitochondrial OXPHOS and MRC have been implicated in the pathogenesis of steatohepatitis.8,25,43. MRC inhibition not only contributes to fat deposition in hepatocytes but it also has the damaging effect of producing ROS. ROS affect the hepatocytes in different ways. ROS trigger the peroxidation of polyunsaturated fatty acids, which in turn activate Kupffer leading to inflammation, and stellate cells, stimulating fibrogenensis.20,44–46 Stress signaling pathways, nuclear and mitochondrial DNA damage, MRC inhibition and cell death are the end results of the modulatory effects of lipid peroxidation.47–49 This cell environment in turn leads to mitochondrial dysfunction, further augmenting the generation of ROS production and induction of cell death, including the activation of inflammatory cytokines such as TNF-α and TGF-β.20,50
The role of adipokinesAdipokines are a host of cytokines derived primarily from the adipose tissue depot. They include leptin, TNF-α, IL-6, adiponectin, resistin, retinol binding protein-4, visfatin.51,52 Most adipokines including tumor necrosis factor-α and resistin, induce insulin resistance and low-grade inflammation through activation of stress-related protein kinases, i.e. c-Jun NH2-terminal kinase-1 (JNK-1), and of the inhibitor kappa kinase beta (IKKβ)/nuclear factor kappa B (NF-κB) pathway.53–55
Leptin considered as an anorexigenic hormone, its levels are elevated in obesity as a result of resistance to its actions.56 Leptin induces dephosphorylation of insulin-receptor substrate 1, rendering hepatocytes more insulin-resistant.56 Leptin is also directly involved in hepatic fibrogenesis through hepatic stellate cells activation.57 Beside Leptin; TNF-α and IL-6 also promote hepatic fibrogenesis.58–60
Adiponectin is a 30 kDa protein abundantly and selectively expressed in white adipose tissue. Binding of adiponectin to its receptors stimulates phosphorylation of AMPK, PPAR activity and fatty acid oxidation in liver.61 Plasma adiponectin was significantly lower in NAFLD patients than controls.62 Adiponectin inhibits liver TNF expression63,64 and also inhibits expression of several cytokines in hepatic stellate cells (HSC).65 Administrations of recombinant adiponectin markedly improved NASH in ob/ob mice and the histological improvement observed in NASH with thiazolidinediones (TZDs) correlate tightly with enhanced adiponectin secretion and adipocyte insulin sensitivity than with visceral fat mass changes, suggesting functional impairment of adipocyte is central to the pathogenesis of NASH.66,67
TNF-α, TNF-regulated cytokines, and cytokines that modulate the synthesis and biologic actions of TNF are produced by many cells within the liver and other organs. These cytokines are the prime mediator of liver injury as well as repair. Thus, antagonism of TNF-α and other injury-related cytokines merits evaluation as a treatment for NASH. In fact, TNF-α inhibition reversed hepatic insulin resistance and liver injury in ob/ob mice and in human NASH.59,68,69
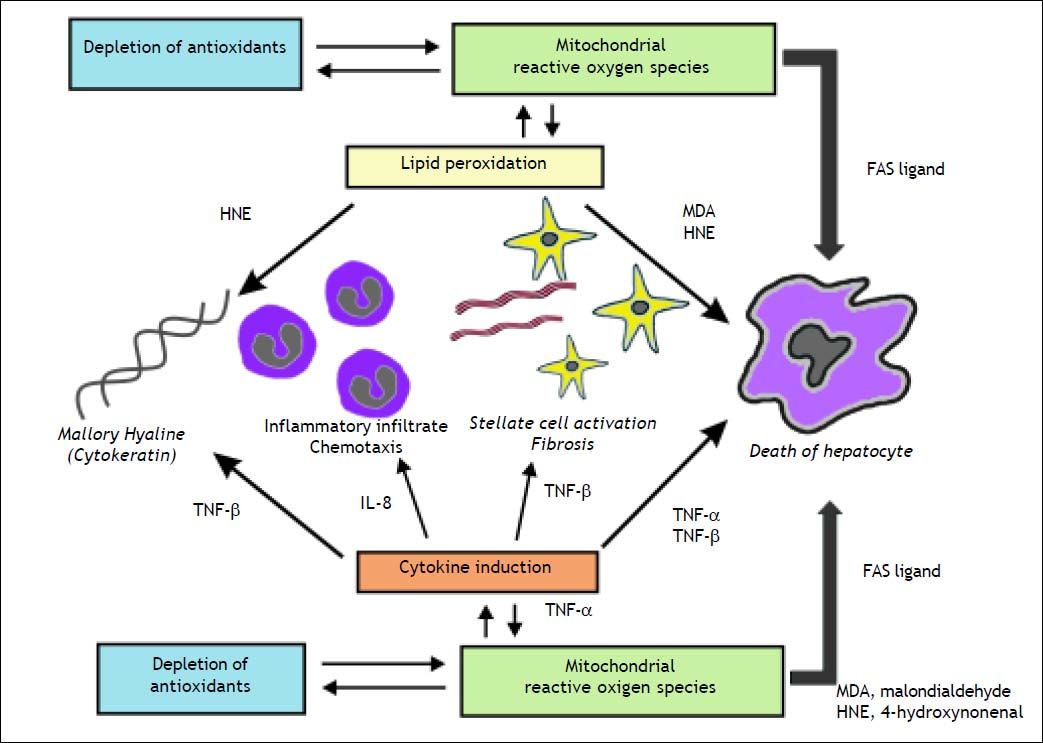
Mitochondrial dysfunction and NASHAccumulating evidence suggests a major role of mitochondrial dysfunction in steatosis and steatohepatitis (Figures 1 and 2).70 Mitochondrial dysfunction not only impairs fat homeostasis in the liver but also leads to overproduction of reactive oxygen species (ROS) that trigger lipid peroxidation, cytokine.70 ROS directly damage mtDNA, respiratory chain polypeptides and mitochondrial cardiolipin. ROS also increase the expression of several cytokines, including transforming growth factor-β (TGF-β), interleukin-8 (IL-8), TNF-α and Fas ligand.13,71 TGF-β, hydroxynonenal (HNE) and IL-8 are chemoattractants for human neutrophils, which may account, in part, for the neutrophil infiltrate.13 TGF-β also induce tissue transglutaminase.13 The induction of tissue transglutaminase by TGF-β could polymerize cytokeratins to generate dense eosinophilic material in the liver cell, the Mallory bodies.71 In patients with NASH, ultrastructural abnormalities of liver mitochondria have been demonstrated.72 In addition, mtDNA is severely depleted in patients with NASH73 and resulting in functional impairment of beta oxidation8 resulting in steatosis.74
; 4 hydroxynonenal (HNE); tumor necrosis factor-α (TNF -α), tumor necrosis factor-β (TNF-β); interleukin-1 (IL-1).")
The figure illustrates the role of mitochondrial reactive oxygen species in the induction of cytokine production and lipid peroxidation leading to the necro-inflammation and fibrosis in non-alcoholic steatohepatitis. Malondiealdehyde (MDA); 4 hydroxynonenal (HNE); tumor necrosis factor-α (TNF -α), tumor necrosis factor-β (TNF-β); interleukin-1 (IL-1).
Normally, hepatocytes express Fas (a membrane receptor), but not Fas ligand, preventing them from killing their neighbors.75 However, several conditions leading to increased ROS formation, such as drugs or alcohol abuse, cause Fas ligand expression by hepatocytes, so that Fas ligand on one hepatocyte can now interact with Fas on another hepatocyte, to cause fratricidal apoptosis.75 Indeed, apoptosis seems to play an important role in NASH.76
The role of microsomes and peroxisomesMembers of the microsomal cytochrome P-450 participate in the generation of oxidative changes in fatty livers via increased production of the free oxygen radical H2O2. Two enzymes, CYP2E1 and CYP4A, are involved in the metabolism of long chain fatty acids (lipo-oxygenation). Over expression of CYP2E1 in the liver has been demonstrated in both animal models of NASH and in patients with NASH.77–79 Insulin resistance and increased cytochrome P450 2E1 (CYP2E1) expression are both associated with and mechanistically implicated in the development of nonalcoholic fatty liver disease. Schattenberg, et al.80 demonstrated that, increased hepatocyte CYP2E1 expression and the presence of steatohepatitis result in the down-regulation of insulin signaling, potentially contributing to the insulin resistance associated with nonalcoholic fatty liver disease. Also, CYP2E1 has a great affinity for electrons and easily forms reactive oxygen species (ROS) that react with the unsaturated bonds of long chain fatty acids and initiate the process of lipid peroxidation.81,82
The enzyme CYP4A is controlled by the transcription factor PPARα, governing genes and is involved in intracellular fatty acid disposal.83,84 Defective states of PPARα or of the peroxisomal boxidation pathway may also play an important role in the development of steatohepatitis.85 It has been shown that mice deficient in PPARα-inducible fatty acid oxidation demonstrate an exaggerated steatotic response to fasting.86 Therefore, in the context of hepatic steatosis, both CYP2E1 and CYP4A could generate the second hit of cellular injury, particularly when antioxidant reserves are depleted.
The role of reduced activity of the MTPIn the endoplasmic reticulum (ER) lumen, MTP lipidates apolipoprotein B (Apo B), to form triglyceride (TG)-rich VLDL particles, which follow vesicular flow to the plasma membrane to be secreted, whereas incompletely lipidated Apo B particles are partly degraded. The functional polymorphism -493 G/T in the MTP gene promoter has been linked to liver disease in NAFLD: GG homozygosity, or carrying a lower MTP activity than the other genotypes, predicted more severe liver histology.87 This polymorphism modulates lipid and lipoprotein levels in healthy and hypercholesterolemic subjects.88,89
Genetic pre-disposition for fatty liver diseaseRecent genome-wide association studies revealed that the genetic variation rs738409 (I148M) in the patatin-like phospholipase 3 gene (PNPLA3) influences NAFLD and plasma levels of liver enzymes,90,91 including predisposition for fibrosis progression.92,93 This polymorphism is a strong predictor of steatosis, inflammation, and fibrosis across different populations, being independent of body mass, insulin resistance, or serum lipid levels.94 The expression of PNPLA3 is regulated by nutrition: fasting inhibits, and high carbohydrate diet feeding increases, PNPLA3 expression.94 PNPLA3 possesses triglyceride hydrolase and DG transacylase activity, and converts lysophosphatidic to phosphatidic acid form.49 By modulating lipid intermediates, dysfunctional PNPLA3 promotes the accumulation of lipotoxic substrates, which lead to lipoapoptosis and inflammation.95 The role of PNPLA3 in the context of drug-induced fatty liver disease is unclear.
Diagnosis of Drug-Induced Fatty Liver DiseaseEstablishing the diagnosis of drug-induced liver injury (DILI), involves a careful evaluation of the temporal profile of drug ingestion and onset of liver disease, the latter developing after and as a consequence of the former.9 In patients with suspected DILI, diagnostic scales, such as the Councils for International Organizations of Medical Sciences/ Roussel Uclaf Causality Assessment Method (CIOMS/RUCAM) scale, may be helpful for the final diagnosis.96 A systematic review of the patient’s medication list must be undertaken, since the list of drugs capable of causing drug-induced fatty liver disease is long. Primary causes of hepatic steatosis/ steatohepatitis are common and must, of course, be ruled out by appropriate imaging and laboratory tests. In particular, drug-induced steatosis and steatohepatitis share many features with alcoholic liver disease, including the presence of significant inflammatory-cell infiltration with ballooning degeneration and the presence of Mallory bodies,97,98 and a careful and accurate history of alcohol consumption must be performed. The case for drug-induced fatty liver disease is strengthened if the reported pattern of injury of the offending drug in the literature is in accordance with the observed clinical and histological picture. The traditional method to evaluate drug-induced phospholipidosis is visual confirmation of myeloid bodies in tissues by electron microscopy and is not routinely performed.
Liver biopsy remains the gold standard for NAFLD diagnosis, however this an invasive technique. Ultrasonography is therefore the recommended first-line imaging technique in clinical practice. There has been intense interest in non-invasive methods to identify advanced fibrosis in patients with NAFLD,99 these include the NAFLD Fibrosis Score,100 enhanced liver fibrosis (ELF) panel100 and transient elastography (fibroscan). The NAFLD fibrosis score is based on six readily available variables (age, body mass index (BMI), hyperglycemia, platelet count, albumin, aspartate aminotransferase (AST)/ alanine aminotransferase (ALT) ratio) and it is calculated using the published formula (http://nafldscore.com). The ELF panel consists of plasma levels of three matrix turnover proteins (hyaluronic acid, tissue inhibitor of metalloproteinases-1 (TIMP-1) and N-terminal propeptide of Type III procollagen (PIIINP) had a 80% sensitivity and 90% specificity for detecting advanced fibrosis.101 Transient elastography is a non-invasive method of assessing liver fibrosis, which can be performed at the bedside or in an outpatient clinic. It employs ultrasound-based technology to measure liver stiffness and has been validated for use in chronic hepatitis C, Human immunodeficiency vius/hepatitis C virus co-infection and cholestatic liver diseases.102 Fibroscan has now been validated in NAFLD103 and represents a useful tool for rapid, non-invasive assessment of liver fibrosis and determining need for biopsy. In 2007, Hamaguchi, et al. generated a scoring system with abdominal ultrasound which could provide accurate information regarding the hepatic steatosis, obesity and the metabolic syndrome in healthy people who do not consume alcohol.104
Drugs that Cause Steatohepatitis and Phospholipidosis IndependentlyThere are three drugs classically identified in the literature to produce drug-induced steatohepatitis with phospholipidosis independently: amiodarone, perhexiline maleate, and DH.7 While phospholipidosis develops after prolonged treatment with these agents in a dose-dependent manner, it does not lead to steatohepatitis. Acute liver injury with microvesicular steatosis mimicking Reye syndrome has been described in the literature.105 Chronic exposure to these agents rarely can also lead to cirrhosis.106,107
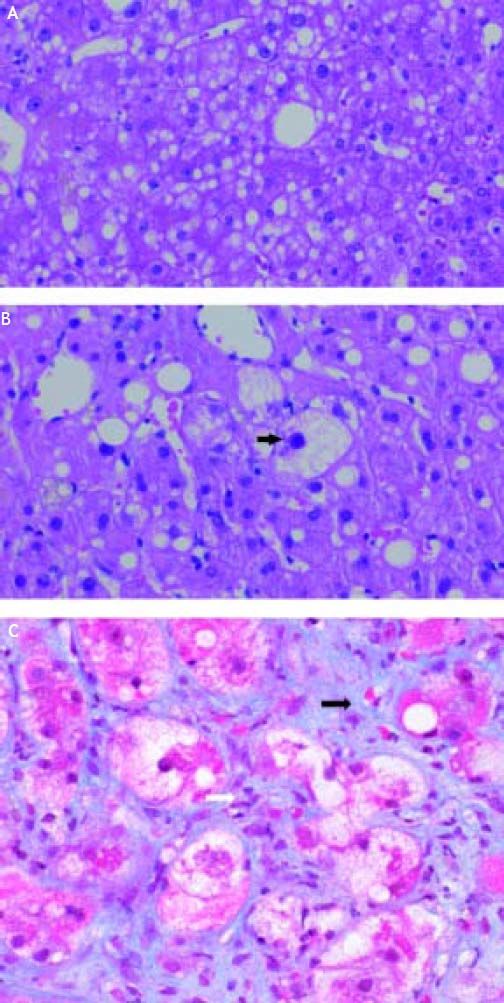
AmiodaroneAmiodarone, an antiarrhythmic drug highly effective for the treatment of atrial and ventricular arrhythmias, is known to have significant thyroid and hepatic side effects. Amiodarone is a hepatic mitochondrial toxin, inhibiting both enzyme complexes in the electron transport chain, as well as adversely affecting β-oxidation, and uncoupling of oxidative phosphorylation. Clinically, amiodarone leads to asymptomatic elevation of serum transaminases in 40-80% of patients being treated with the drug, which can be associated with mild cholestasis.108 Amiodarone toxicity, on the other hand, develops in a much smaller fraction of patients, 1-3%; however, when it occurs, it can be severe, histologically characterized by steatohepatitis resembling alcoholic hepatitis with Mallory bodies, ballooning degeneration, and inflammatory PMN infiltrates (Figure 3),97 and less frequently microvesicular steatosis.105 The liver injury associated with amiodarone toxicity typically resolves with discontinuation of the drug,6,109 however, full recovery may be a protracted process over the course of weeks to months secondary to the drug’s long half-life.110
 secondary to amiodarone toxicity. Original magnification 400x. C. Trichrome stain demonstrating peri-sinusoidal fibrosis (solid arrows) secondary to steatohepatitis. Note the fibrous expansion of portal fields assuming the appearance of stellatefibrosis, as well as inflammatory infiltrate (white arrow). Trichrome stain. Original magnification 400x. Photographs provided by Pamela B. Sylvestre, M.D., affiliated with the University of Tennessee/Methodist University Hospital Transplant Institute, Memphis, TN.")
A. Microvesicular steatosis secondary to amiodarone toxicity. The majority of the lipid droplets are the same size or smaller than the nuclei of the hepatocytes. Original magnification 400x. B. Steatohepatitis with ballooning degeneration and a Mallory-Denk body (arrow) secondary to amiodarone toxicity. Original magnification 400x. C. Trichrome stain demonstrating peri-sinusoidal fibrosis (solid arrows) secondary to steatohepatitis. Note the fibrous expansion of portal fields assuming the appearance of stellatefibrosis, as well as inflammatory infiltrate (white arrow). Trichrome stain. Original magnification 400x. Photographs provided by Pamela B. Sylvestre, M.D., affiliated with the University of Tennessee/Methodist University Hospital Transplant Institute, Memphis, TN.
Perhexiline maleate is a cationic amphiphile antianginal drug previously used extensively in Europe. Its hepatic toxicity is similar to amiodarone, histologically mimicking alcoholic steatohepatitis, associated with phospholipidosis.111,112 Pharmacologically, the drug impairs hepatic liver metabolism similarly to amiodarone, concentrating in the mitochondria, uncouples oxidative phosphorylation and inhibits ß-oxidation of fatty acids. Perhexiline maleate also decreases the exit of triglycerides from the liver, resulting in significant microvesicular steatosis, necrosis, and inflammation.22 Perhexiline is catabolized by cytochrome P-450 2D6; 3-7% of Caucasians are slow metabolizers for perhexiline; such individuals are at a greater risk for steatohepatitis and neuropathy, another side effect of the drug. Perhexiline half-life is long, 3 to 12 days, resulting in delayed hepatic clearance, particularly in slow metabolizers.113 This drug is however is no longer in the United States market although continue to be used in some countries like Australia and New Zealand.
Diethylaminoethoxyhexestrol (DH)DH is a coronary vasodilator, antianginal cationic amphiphile drug formerly used extensively in Japan.7 Similar to perhexeline maleate, it induces phospholipidosis and a histological picture very similar to alcoholic steatohepatitis. This drug is concentrated to high levels in hepatocyte lysosomes, inhibiting phospholipase A1.114 Similar to amiodarone and perhexeline, steatohepatitis is a consenseque of DH’s accumulation in mitochondria, where it inhibits mitochondrial respiration, causing depletion of ATP, inhibition of β-oxidation, and consequently, lipid peroxidation.115
Drugs that can Precipitate Hepatic Steatosis/SteatohepatitisTamoxifen, an antiestrogenic drug widely used for the primary and secondary prevention of breast carcinoma is commonly associated with hepatic steatosis,116,117 though rarely with steatohepatitis118,119 or cirrhosis.120,121 The mechanism of tamoxifen-associated steatohepatitis has not been extensively studied, but it is postulated that its effect may be secondary to its estrogen agonist activity and inhibition of mitochondrial β-oxidation of fatty acids.122,123 Tamoxifen often increases serum triglycerides, another risk factor for NASH,124 and it is particularly has been found to cause steatohepatitis in patients with pre-existing liver steatosis, especially patients with high BMIs, hyperglycemia and hyperlipidemia. However, an estrogen-receptor (ER) independent, direct effect of tamoxifen on the mitochondrial respiration rate and phosphorylation efficiency has been also described.125 Histologically, tamoxifen-induced steatohepatitis mimics alcoholic steatohepatitis.118
Raloxifene, a selective estrogen receptor modulator (SERM) used in the treatment and prevention of osteoporosis in postmenopausal women, has also rarely been described in the literature as aggravating steatosis in patients with underlying non-alcoholic fatty liver disease,126 in a rat model, raloxefine inhibited both mitochondrial and peroxisomal β-oxidation.127
Similar to estrogens, glucocorticoids frequently cause hepatic steatosis, though rarely cause steatohepatitis.128In vitro dexamethasone has been shown to inhibit mitochondrial ß-oxidation, decrease hepatic triglyceride secretion resulting in microvesicular steatosis in a mouse model.129 While the exact mechanism by which steatosis evolves into steatohepatitis in certain patients receiving glucocorticoids remains undetermined, plausibly it may involve the exacerbation of metabolic mechanisms known to contribute to NASH, such as insulin insensitivity, and hypertriglyceridemia.7
VPA and carbamazepene (CBZ), are widely used antiepileptic drugs. VPA in particular has well-established, common metabolic side-effects including weight gain130 and increased insulin resistance.131 VPA, and to a lesser degree CBZ, have been associated with increased risk for ultrasonographic signs of fatty liver disease.132 CBZ-induced steatosis/steatohepatitis may be idiosyncratic; while hepatotoxicity associated with CBZ is well described in the literature, typically causing hepatitis,133,134 steatosis/steatohepatitis is rare.135
Methotrexate, an antimetabolite drug used as a chemotherapeutic agent as well as in the treatment of autoimmune disorders, is known to cause steatohepatitis, particularly in patients with underlying cardiometabolic risk factors such as diabetes, obesity, and heavy ethanol use, accelerating liver injury caused by NASH in a dose-dependent manner.136 Chemotherapy-associated steatohepatitis has also been described in patient undergoing chemotherapy with agents such as was irinotecan and oxaliplatin, particularly in patients with underlying obesity.137 Irinotecan has also been shown in a large study to be associated with steatohepatitis regardless of body mass index (BMI), though clinically significant outcome, such as perioperative mortality, appears to be linked rather to BMI.138 Some authors advocate for further studies in this area are to support this association, and to determine if chemotherapy-associated steatohepatitis could be modified by drug or dose adjustment.139
The nucleoside reverse transcriptase inhibitors (NRTI) zidovudine and stavudine, commonly used in the treatment of HIV, have also been linked to macrovacuolar and microvesicular steatosis and severe lactic acidosis,140,141 indirectly via the inhibition of mitochondrial DNA polymerase142 or through direct inhibition of fatty acid oxidation,143,144 In addition, NRTIs are associated with insulin resistance145 and may be an independent risk factor for the development of hepatic steatosis according to a large crosssectional study,146 though this is debated.147 Treatment with protease inhibitors (PIs) such as ritonavir have been associated with lipodystrophy148,149 which is in turn associated with insulin resistance and hyperlipidemia.150
Management of Drug-Induced Fatty Liver DiseaseDespite an increased understanding of the pathogenesis of fatty liver disease, there are few effective therapies available. Current treatment strategies for fatty liver disease in general are primarily directed towards improvement of metabolic parameters which contribute to disease pathogenesis, including weight loss, exercise, lipid and glycemic control.98 It is unclear if lifestyle modification strategies, such as exercise and weight loss, or pharmacotherapy will have any beneficial role in the management of druginduced steatohepatitis considering the cardio metabolic risk factors and insulin resistance are not the key player in its pathogenesis. Particular to drug-induced fatty liver disease, the identification and removal of the culprit drug is key, and the mainstay of treatment.
Vitamin E, has been shown to improve liver histology in non-diabetic adults with biopsy-proven NASH and therefore according to AASLD guidelines is recommended as first-line pharmacotherapy in pa- tient with primary NASH.151 Vitamin E (alfa-tocopherol) is relatively safe and readily available. The rationale for using vitamin E in patients with NASH is derived mostly from its antioxidant properties, and could potentially be useful in drug-induced steatohepatitis. Considering the rarity of the cases of drug-induced fatty liver disease, a large scale randomized trial is unlikely to be available soon.
A small open label pilot study has generated some interest in using bezafribate in preventing progression NASH induced tamoxifen in breast cancer patients. The study however used liver/spleen CT ratio for follow up as a surrogate to liver biopsy, therefore needs further evaluation in better designed study prior to any recommendation for use.152 Despite this is a small study, such an approach for prevention of progression related to drug induced NASH needs to be evaluated with potential promising drugs.
Possible beneficial effects of other anti-oxidants such as S-adenosylmethionine (SAMe),153 and Silymarin154 has been speculated but lacks confirmation. Obeticholic acid (6α-ethyl-chenodeoxycholic acid) is a semisynthetic derivative of the primary human bile acid chenodeoxycholic acid, the natural agonist of the farnesoid X receptor, which is a nuclear hormone receptor that regulates glucose and lipid metabolism. A recently concluded, multicentre, double-blind, placebo-controlled, parallel group, randomized clinical trial obeticholic acid was found to significantly improve the histological features of non-alcoholic steatohepatitis, but its long-term benefits and safety need further clarification. These and many new drugs under development (details beyond the scope of this review), certainly has generated enormous interest and hope in the treatment of NAFLD, and NASH in particular in the last few years, some of them could potentially be used in drug-induced steatohepatitis as well.155
ConclusionTrue drug-induced steatohepatitis is rare, and only unequivocally associated with three drugs: amiodarone, perhexiline, and DH, all which share a common hepatotoxicity mechanism involving mitochondrial ATP formation and fatty acid oxidation. Several other drugs, such as tamoxifen, methotrexate and irinotecan have been implicated in the exacerbation of steatohepatits/steatosis in the background setting of pre-existing cardiometabolic risk factors, especially obesity. Other drugs, such as steroids and antiepileptic agents may lead to steato- hepatitis by indirect induction of metabolic risk factors for NASH, such as insulin insensitivity, and hypertriglyceridemia. Optimal treatment of drug-induced steatohepatitis is unclear. Further research is needed to determine the exact mechanisms by which drug-induced steatosis progresses into steatohepatitis and fibrosis, perhaps leading to tailored modalities for the treatment and prevention of NASH.
Author DeclarationThis manuscript is not being considered for publication elsewhere.
Abbreviations- •
ALT: alanine aminotransferases.
- •
AMPK: AMP-activated protein kinase.
- •
Apo B: apolipoprotein B.
- •
AST: aspartate aminotransferases.
- •
ATP: adenosine triphosphate.
- •
AZT: azidothymidine.
- •
BMI: body mass index.
- •
CADs: cationic amphiphilic drugs.
- •
CBZ: carbamazepine.
- •
CIOMS: Councils for International Organizations of Medical Sciences.
- •
D4T: stavudine.
- •
DDI: didanosine.
- •
DH: 4,4’-diethylaminoethoxyhexestrol.
- •
DILI: drug-induced liver injury.
- •
DIPL: drug-induced phospholipidosis.
- •
ELF: enhanced liver fibrosis.
- •
ER: endoplasmic reticulum.
- •
FAO: fatty acid oxidation.
- •
FFAs: free fatty acids.
- •
HNE: hydroxynonenal.
- •
HSC: hepatic stellate cells.
- •
IKKβ: inhibitor kappa kinase beta.
- •
IL-8: interleukin-8.
- •
LCFAs: long chain fatty acids.
- •
MRC: mitochondrial respiratory chain.
- •
mtDNA: mitochondrial DNA.
- •
MTP: microsomal triglyceride transfer protein.
- •
NAFLD: non-alcoholic fatty liver disease.
- •
NAS: NAFLD acitivity score.
- •
NASH: non-alcoholic steatohepatitis.
- •
NF-kB: nuclear factor kappa B.
- •
NHANES: National Health and Nutrition Examination Surveys.
- •
NRTI: nucleoside reverse transcriptase inhibitors.
- •
cNSAIDs: non-steroidal anti-inflammatory drugs.
- •
OXPHOS: oxidative phosphorylation.
- •
PIIINP: N-terminal propeptide of type III procollagen.
- •
PNPLA3: patatin-like phospholipase 3 gene.
- •
RNS: reactive nitrogen species.
- •
ROS: reactive oxygen species.
- •
RUCAM: Roussel Uclaf Causality Assessment Method.
- •
SERM: selective estrogen receptor modulator.
- •
TG: triglyceride.
- •
TGF-β: transforming growth factor-ß.
- •
TIMP-1: tissue inhibitor of metalloproteinases-1.
- •
TNF-α: transforming growth factor-α.
- •
TZDs: thiazolidinediones.
- •
VLCFAs: very long chain fatty acids.
- •
VLDL: very low-density lipoprotein.
- •
VPA: valproic acid.



