



Metabolic dysfunction-associated steatotic liver disease (MASLD) is one of the primary causes of chronic liver disease and may lead to liver cirrhosis and hepatocellular carcinoma. Recent reports suggested that DEAD-box RNA helicase (DDX3) acts as a sensor of free fat accumulation and may modulate the pathogenesis via miRNAs. Hence, we hypothesized that DDX3 might modulate MASLD progression via miRNA-141-mediated inhibition of Sirt-1 and autophagy.
Materials and MethodsRNA and total protein were isolated from free fatty acid-treated HepG2 cells or CDAA-fed C57BL/6 mice (6 mice per group) for 6, 18, 32, or 54 weeks. The cells were transfected with DDX3 or miR-141 or siRNA to DDX3, and Western blots for autophagy markers were performed.
ResultsThe FFAs induced the DDX3 and miRNA-141 expression, while downregulating Sirt-1, beclin-1, Atg7, and LC3-II. Overexpression of DDX3 resulted in increased miRNA-141. Overexpression of DDX3 or miRNA-141 downregulated Sirt-1 expression and autophagy marker proteins, while these effects were reversed with siRNA to DDX3. The expression of both DDX3 and miRNA-141 was significantly increased, while autophagy markers were downregulated in CDAA-fed mice.
ConclusionsThese results confirmed that FFA-induced DDX3 induced the expression of miRNA-141, which in turn targeted Sirt-1 and decreased autophagy.
Metabolic dysfunction-associated steatotic liver disease (MASLD), previously known as non-alcoholic fatty liver disease (NAFLD), has emerged as a burgeoning public health concern, escalating to become one of the prime contributors to chronic liver disease worldwide [1,2]. Despite its rapidly increasing prevalence, the intricate molecular mechanisms underlying the progression of MASLD remain unclear. Recent findings suggest that there is a pivotal role of DEAD-box RNA helicase 3 (DDX3), a member of the RNA-binding protein family, in serving as a sentinel for the accumulation of free fat within the liver [3,4]. DDX3 has also been shown to modulate several cellular functions such as cell cycle, apoptosis, viral replication and tumorigenesis [3]. DDX3 protein also could interact with various human and viral proteins and binds with RNA [5,6]. It was shown to have an oncogenic role in tumorigenesis by promoting transformation, invasion and cell growth [7–9]. Several studies also have tried to use this protein as a therapeutic target for the development of anticancer drugs due to its role in oncogene in various type of cancer [7,10,11]. The functions of this gene as an oncogene or tumour suppressor remain unclear. DDX3 translocates from the nucleus to the cytoplasm and interacts with exportin 1, regulating HCV and HIV replication [12]. DDX3 interacts with influenza A virus NS1 and NP proteins, regulating stress granule formation and IFN-β promoter transcription [3,13]. Beyond this, it's been found that DDX3 has role in driving inflammasome activation [14]. However, the activation mechanism of DDX3, such as protein modification, remains to be elusive. DDX3, beyond its conventional functions, has been shown to be involved in regulating microRNA (miRNA) biogenesis, facilitating the transformation of pre-miRNAs into mature miRNAs [15–17]. Although DDX3 is expressed aberrantly in various types of cancers, its interaction with cellular components also has a regulatory effect on the tumorigenic process [18]. But, it's not clear whether DDX3 involves any miRNA in the pathogenesis of MASLD. microRNAs are non-coding single-stranded RNAs of around 22 nucleotides that play a variety of physiological roles, including cell proliferation, survival, metabolism, differentiation, and invasion [19]. MicroRNAs are transcribed by RNA polymerases II and III, which produce precursors that are then cleaved to make mature microRNAs [20]. MicroRNAs downregulate gene expression at the post transcriptional level by targeting and binding with 3′ untranslated region (3′UTR) of specific messenger RNA [21]. miRNAs have epigenetic mechanisms that play a major role in the progression of MASLD to its more severe stages [22]. A recent study showed that miR-141 directly targets Sirtuin-1/AMP-activated protein kinase pathways to control hepatic steatosis in hepatocytes [23]. On the other hand, DDX3 has a major role in biogenesis and modulation of different deregulated hepatic microRNAs and may modulate the progression of MASLD. But the exact mechanism is yet to be revealed. It might be possible that DDX3 may exert a significant influence on the progression of MASLD by orchestrating miRNA-mediated regulatory pathways, particularly those targeting autophagy, a cellular process known to be vital in the maintenance of liver health [15,24]. In this study, we showed that DDX3 is involved in inhibiting autophagy via miRNA-141-mediated Sirt-1 downregulation in CDAA-induced MASLD mice model system.
2Materials and Methods2.1Cell culture and free fatty acid (FFA) treatmentHuman hepatic HepG2 cells were cultured and maintained in complete DMEM media (HiMedia Laboratories, Mumbai, India, Cat No AL007G) with the addition of 10 % FBS (HiMedia Laboratories) and 1 % penicillin-streptomycin antibiotic (Gibco, Thermo Fisher Scientific, MA, USA) at 37 °c in the presence of 5 % CO2. The cells (2 × 105 cells per well) were plated in 6 well plates and after 24 h, 650 μM each of oleic acid (OA; Calbiochem, Cat. No 4954), palmitic acid (PA; Calbiochem, Cat No 506,345) or linoleic acid (LA; Calbiochem, Cat No 436,305) were added to the cells and incubated for 48 h and 72 h to isolate RNA or protein, respectively.
2.2Transfection experimentsHepG2 cells were transfected with miR-141 premiRs (Sigma-Aldrich, St. Louis, MO, USA), siDDX3 siRNAs (Thermo Scientific, Carlsbad, USA; Cat No 145,804), non-specific miRNA (NS; Sigma-Aldrich (St. Louis, MO, USA) or DDX3 expression plasmid. The transfection was done using siPORT™ NeoFX™ transfection reagent (Thermo Fisher, Carlsbad, CA, USA), following the manufacturer's protocol. Forward transfection was done with lipofectamin 2000, as described by us earlier [15]. After the transfection, the cells were incubated in complete media for up to 48 h or 72 h, and the cells were collected for the isolation of RNA or protein, respectively.
2.3CDAA-induced MASLD mice model systemAnimal Ethics Committee of South Asian University, New Delhi, India, approved all the in vivo experiments used in this study. Three groups of 5–6 weeks old male C57BL/6 mice (6 mice per group) were fed with normal chow diet (sham-operated control), choline sufficient L-amino acid defined diet (CSAA; Dyets Inc. Cat. No 518,754) or choline-deficient L-amino acid defined diet (CDAA; Dyets Inc. Cat. No 518,753) for 6, 18, 32, and 54 weeks with free access of diet and water. At the end of feeding, the mice were euthanized, and the livers were carefully dissected. The liver tissues were then sliced into smaller pieces and promptly frozen by snap-freezing method after quick washing with PBS (phosphate-buffered saline). Later, they were stored at −80 °C until use. They were utilized to isolate total RNA and total protein. The establishment of the MASLD mice model was confirmed by histological analysis of liver tissue measurement of TG and HbA1c, which was described in our previous study [25].
2.4Total RNA isolation, cDNA synthesis and RT PCRThe total RNA from HepG2 cells or from the homogenized mice liver tissue was prepared using Trizol reagent following the manufacturer's instructions (Thermo Fisher, Cat. No 15,596,026). cDNA was prepared from these RNA through reverse transcription using the miRCURY LNATM RT Kit (Qiagen, Maryland, USA, Cat No 339,340). Real-time PCR was done to analyze the expression levels of miR-141 or 5S rRNA using the SYBR green master mix (Qiagen). The PCR was done using the ViiA 7 Real-Time PCR system (Applied Biosystems, Thermo Fisher Scientific). The 2-ΔΔCT was calculated for each experiment and then the relative expression was plotted by keeping control as 1-fold.
2.5Western blotting experimentsMammalian protein extraction reagent, MPER (Thermo Fisher, Cat No 78,501), was used to extract proteins from HepG2 cells. To prevent protein degradation, a protease inhibitor (PI) cocktail (Thermo Fisher, Cat No 78,430) was added to the MPER reagent in a 100:1 ratio. The mixture was then added to six-well plates containing the HepG2 cells and allowed to incubate for 10 min on ice. The lysed cells were sonicated at a low temperature to avoid protein degradation. After centrifugation, the protein was collected for further experiments. Protein from frozen mice liver tissues was also isolated following same technique after crushing the tissues properly using mechanical method. For western blot analysis, 40–80 μg of protein was loaded on 8–15 % SDS-polyacrylamide gel electrophoresis depending on protein size, transferred to PVDF membranes and blocked using 5 % non-fat dry milk. The membranes were incubated with primary antibodies to DDX3, Sirt-1, ATG7, beclin-1, LC3-II and β-actin. In all the Western blot results, β-actin was used as a loading control, and the relative expression was calculated by dividing the gene of interest by β-actin expression. After washing with PBS-T, the blots were further incubated with specific HRP conjugated secondary antibodies. The membranes were washed and developed using ClarityTM Western ECL Substrate (Bio-Rad, Cat. No 170–5061). The band intensities were quantified using ImageJ software (NIH, USA).
2.6Statistical analysisAll the in vitro experiments were conducted three times in duplicates. All in vivo data (6 mice per group) was collected, analyzed and plotted. As a loading control β-actin was used in all the Western blot experiments and 5S rRNA was used as internal control in all the real time PCR experiments. The Western blot results were quantified using Image J software and the mean ± SD was calculated. The relative expression was calculated by dividing gene of interest with β-actin. To determine the significant differences between the two groups, an unpaired Student's t-test was performed and one-way ANOVA was performed to compare more than two groups. A difference was considered statistically significant only when the p-value was less than or equal to 0.05.
2.7Ethical statementThe in vivo mice work was approved by the Institutional Animal Ethics Committee, South Asian University, New Delhi.This placement ensures consistency with standard formatting practices and clarity for readers.
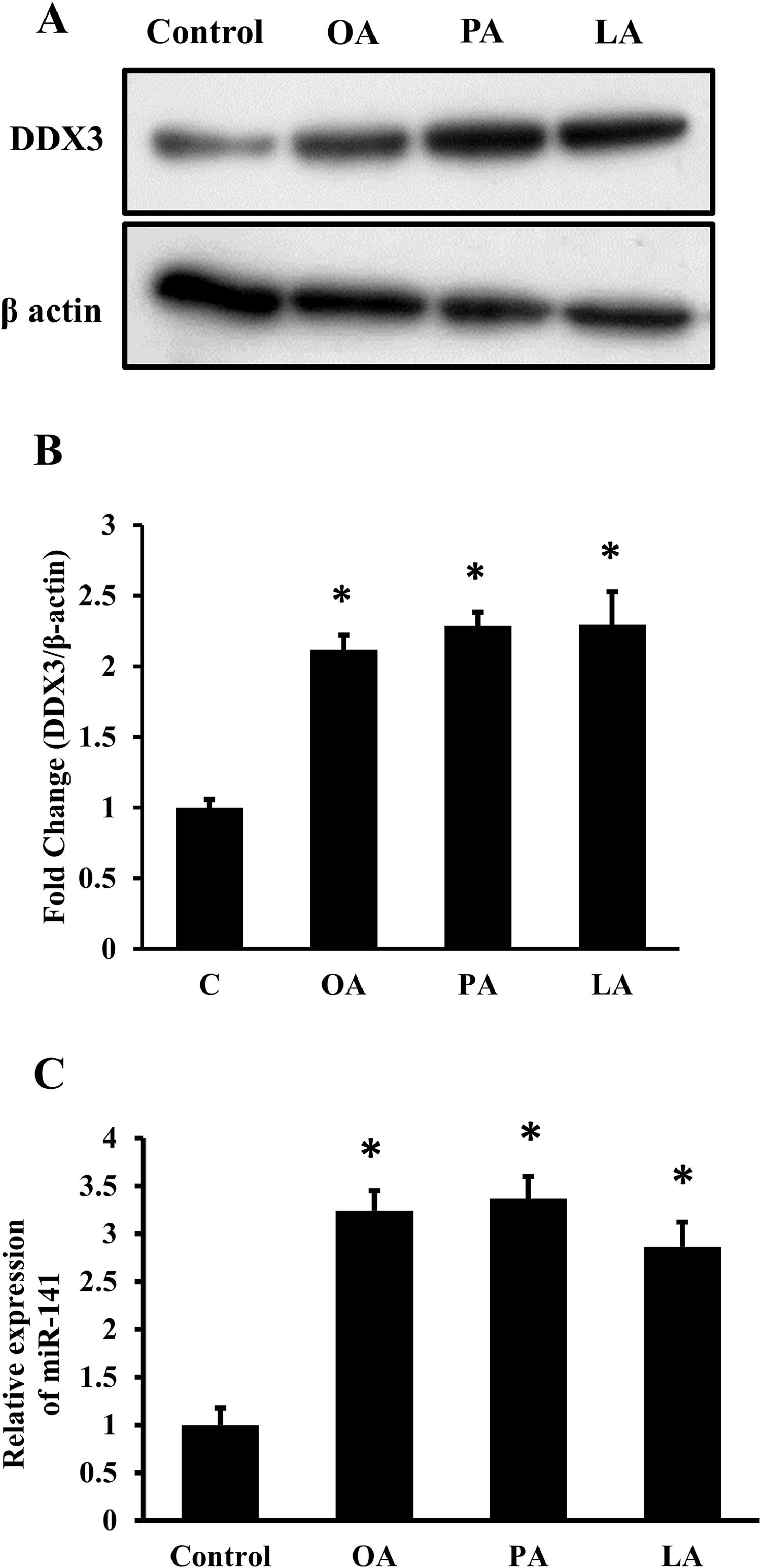
3Results3.1FFAs induce DDX3 expression and miR-141 in hepatic cellsDDX3 is shown to be modulating cellular functions in various cell types. To study the effect of free fatty acids on the expression of DDX3, HepG2 cells were incubated with OA, PA, and LA (650 μM each) for 72 h and found that there was a significant increase in the expression of DDX3 (Fig. 1A). The band intensities were quantified using Image J software, and a two-fold increase in the DDX3 expression was found (Fig. 1B). Next, total RNA was isolated from the FFA-treated cells and the real time PCR results showed that there was a significant increase in the expression of miR-141 in FFA-treated HepG2 cells (Fig. 1C) and the normalization data showed that there was a 3-fold increase in the expression of miR-141 (Fig. 1C).

FFAs induced DDX3 and miRNA-141 expression. HepG2 cells were incubated with 650 μM each of OA, PA, LA. Cells were collected after 48 or 72 h for RNA or protein isolation, respectively. A. The Western blots were performed using total cellular protein to determine the expression of DDX3 and β-actin. B. The band intensities were quantified using Image J software and the fold change was calculated C. The cells were incubated with FFAs for 48 h and the total RNA was isolated. cDNA was synthesized and the real-time PCR for miRNA-141 was performed. The 2-ΔΔCT was calculated and then the relative expression was plotted by keeping control as 1-fold. The mean ±SD was calculated from 3 experiments in duplicates, and the Student's t-test was performed between the control and the FFA-treated cells (n = 3; *p < 0.05).
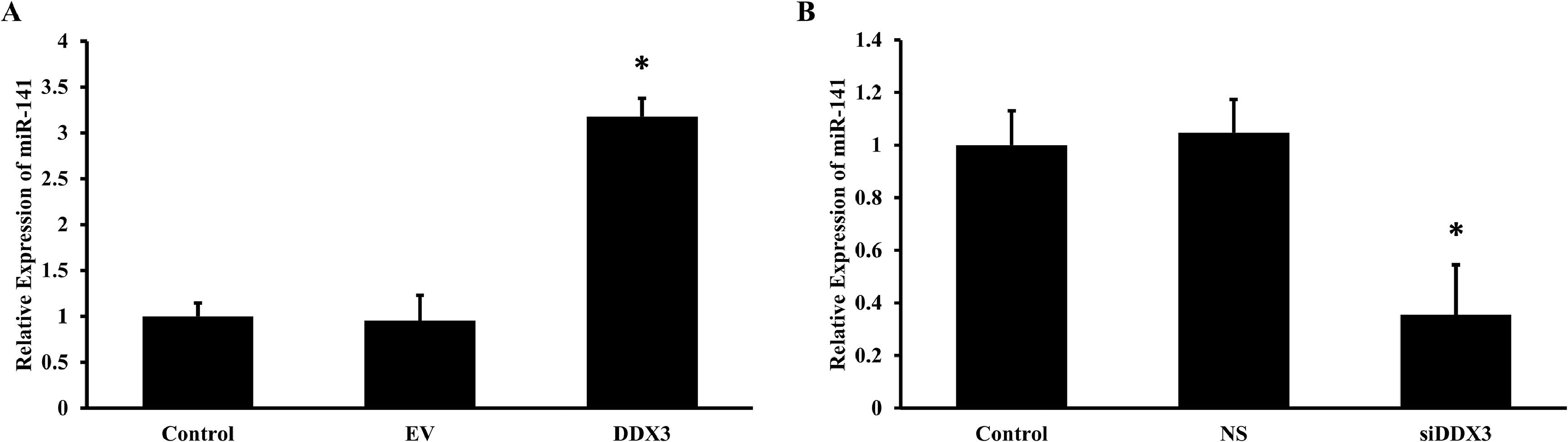
Previous studies have shown that DDX3 plays an important role in miRNA biogenesis. Previously, we had shown that a novel role of DDX3 in upregulating miR-34 [15]. To study the effect of DDX3 on the expression of miR-141, DDX3 was overexpressed in HepG2 cells, and the real time PCR showed that there was a 3-fold increase in the expression of miR-141, compared to the control or empty vector-transfected cells (Fig. 2A) (n = 3; *p < 0.05). Next, the DDX3 was inhibited using siRNAs to DDX3 and the expression of miR-141 was determined in HepG2 cells. The results showed that there was a significant 60 % inhibition in the expression of miR-141 in the siRNA to DDX3-transfected cells (Fig. 2B) (n = 3; *p < 0.05).
 (A) or siRNAs to DDX3 or non-specific RNAs (NS) (B) for 48 h. After incubation, RNA was isolated and real-time PCR for miRNA-141 was performed. All the experiments were repeated 3 times in duplicates. The mean ±SD was calculated and the Student")
DDX3 upregulates the expression of miRNA-141. HepG2 cells were cultured and transfected with either plasmids containing DDX3 or empty vector (EV) (A) or siRNAs to DDX3 or non-specific RNAs (NS) (B) for 48 h. After incubation, RNA was isolated and real-time PCR for miRNA-141 was performed. All the experiments were repeated 3 times in duplicates. The mean ±SD was calculated and the Student's t-test was performed between the control and the transfected cells (n = 3; *p < 0.05).
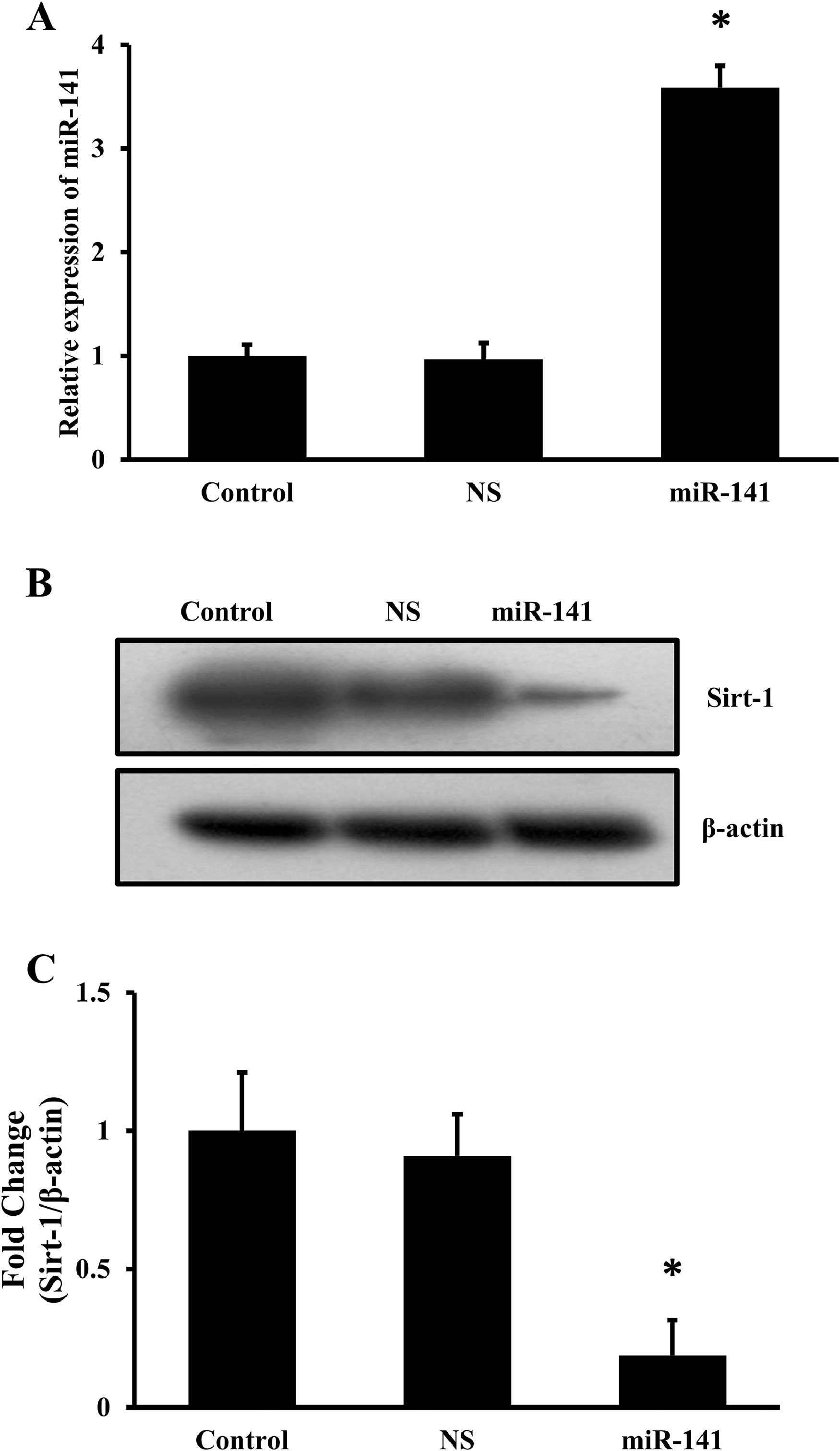
Using bioinformatic approach, the target for miR-141 was determined and found Sirt-1 was its primary target. Previous studies also showed that Sirt-1 is the target for miR-141 [23]. Hence, premiR-141 was transfected in HepG2 cells, and the expression of miR-141 was determined. It was found that there was a 3.5-fold increase in miR-141 expression (Fig. 3A). Next, the total cellular protein was isolated from the control, non-specific miRNA- and miR-141 premiRs-transfected HepG2 cells and Sirt-1 expression was determined by Western blots. It was found that there was a significant downregulation of Sirt-1 (Fig. 3B) and the quantitation of the band intensities showed that there was an 80 % decrease in the Sirt-1 expression (Fig. 3C).
. B. After 72 h, Western blots for Sirt-1 and β-actin was determined. C. The band intensities were quantified using Image J software. The mean ± SD was calculated from 3 experiments in duplicates, and the Student")
miRNA-141 targets Sirt-1. HepG2 cells were transfected with miRNA-141 premiRs or NS for 48 or 72 h. A. After 48 h, expression of miRNA-141 and 5S rRNA was determined (n = 3; *p < 0.05). B. After 72 h, Western blots for Sirt-1 and β-actin was determined. C. The band intensities were quantified using Image J software. The mean ± SD was calculated from 3 experiments in duplicates, and the Student's t-test was performed between the control and the transfected cells (n = 3; *p < 0.05).
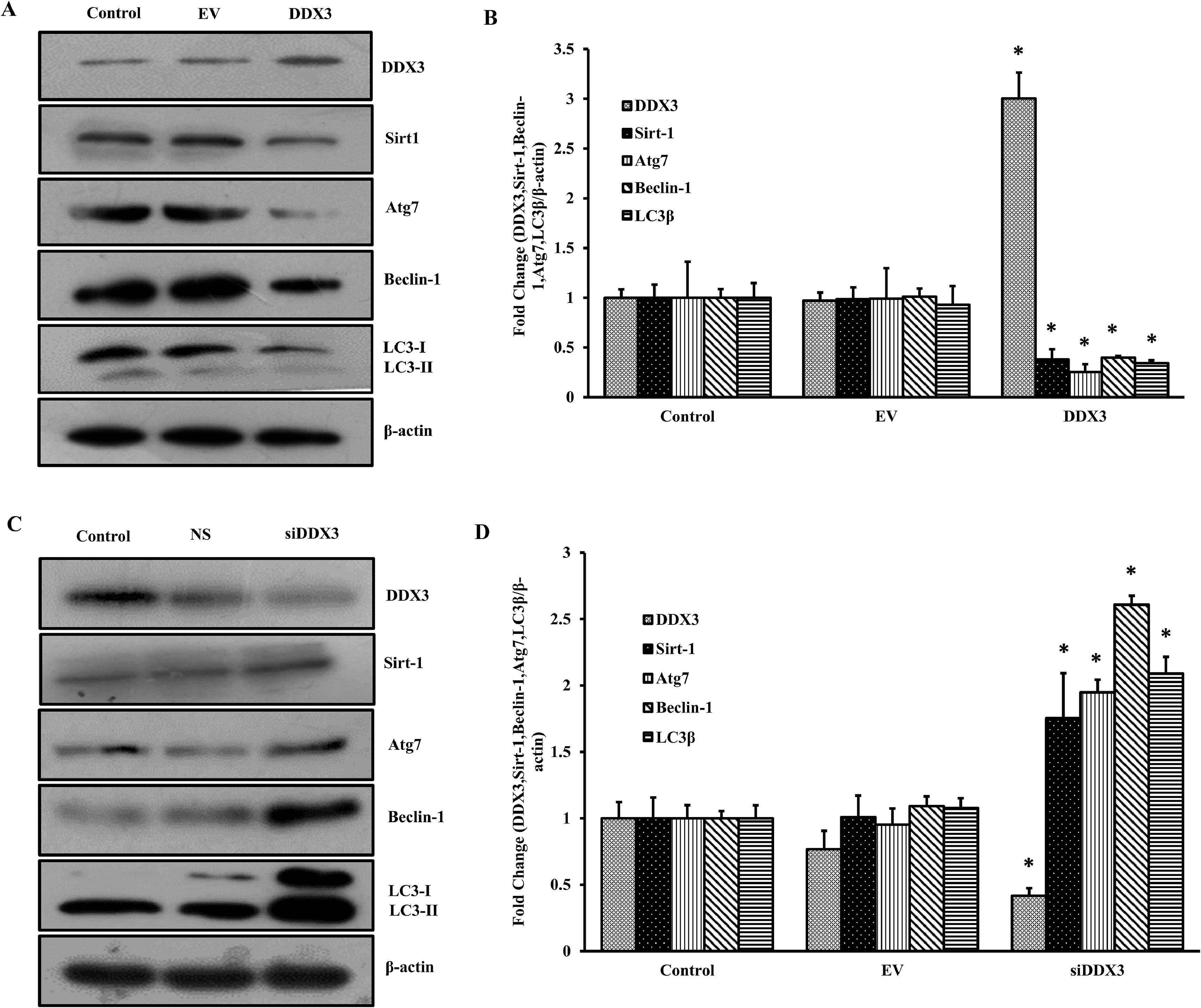
HepG2 cells were transfected with the DDX3 containing expression plasmids. After 72 h, the Western blots were performed for the expression of DDX3, Sirt-1, Atg7, beclin-1, LC3-II and β-actin. The expression of DDX3 was significantly increased in the cells overexpressing DDX3 compared to control or empty vector transfected cells. On the other hand, expression of the autophagy marker proteins was significantly decreased (Fig. 4A). The band intensities were quantified using Image J software and found that there was a 3-fold increase in the DDX3 expression, while the expression of autophagy marker protein was decreased by 50–60 % in comparison to control or empty vector transfected cells (Fig. 4B). To confirm the findings, the cells were incubated with siRNAs in DDX3, where non-specific RNAs were used as a control. The results showed that there was a significant inhibition of DDX3 expression, whereas the expression of autophagy marker proteins was increased significantly (Fig. 4C). The band intensities showed that there was a 50 % decrease in the expression of DDX3, while the expression of autophagic proteins was increased by at least 2–2.7-fold compared to control or NS-miRNA transfected cells (Fig. 4D).

DDX3 inhibits autophagy. HepG2 cells were transfected with plasmids containing DDX3, EV, siRNAs to DDX3 or NS for 72 h. Total cellular protein was isolated from HepG2 cells and Western blots were done for the autophagy marker proteins. A. Representative blots showing the expression of DDX3, Atg7, beclin-1, LC3-II and β-actin in DDX3 transfected cells. B. The band intensities were quantified and the mean ± SD was calculated from 3 experiments in duplicates, and the Student's t-test was performed between the control and the transfected cells (n = 3; *p < 0.05). One-way ANOVA was performed to check the significant difference among these groups (n = 3; *p < 0.05). C. Representative blots showing the expression of autophagy marker proteins in NS or DDX3 siRNAs transfected cells. D. The band intensities were quantified and the mean ± SD was calculated from 3 experiments in duplicates, and the Student's t-test was performed between the control and the transfected cells (n = 3; *p < 0.05). One-way ANOVA was performed to check the significant difference among these groups (n = 3; *p < 0.05).
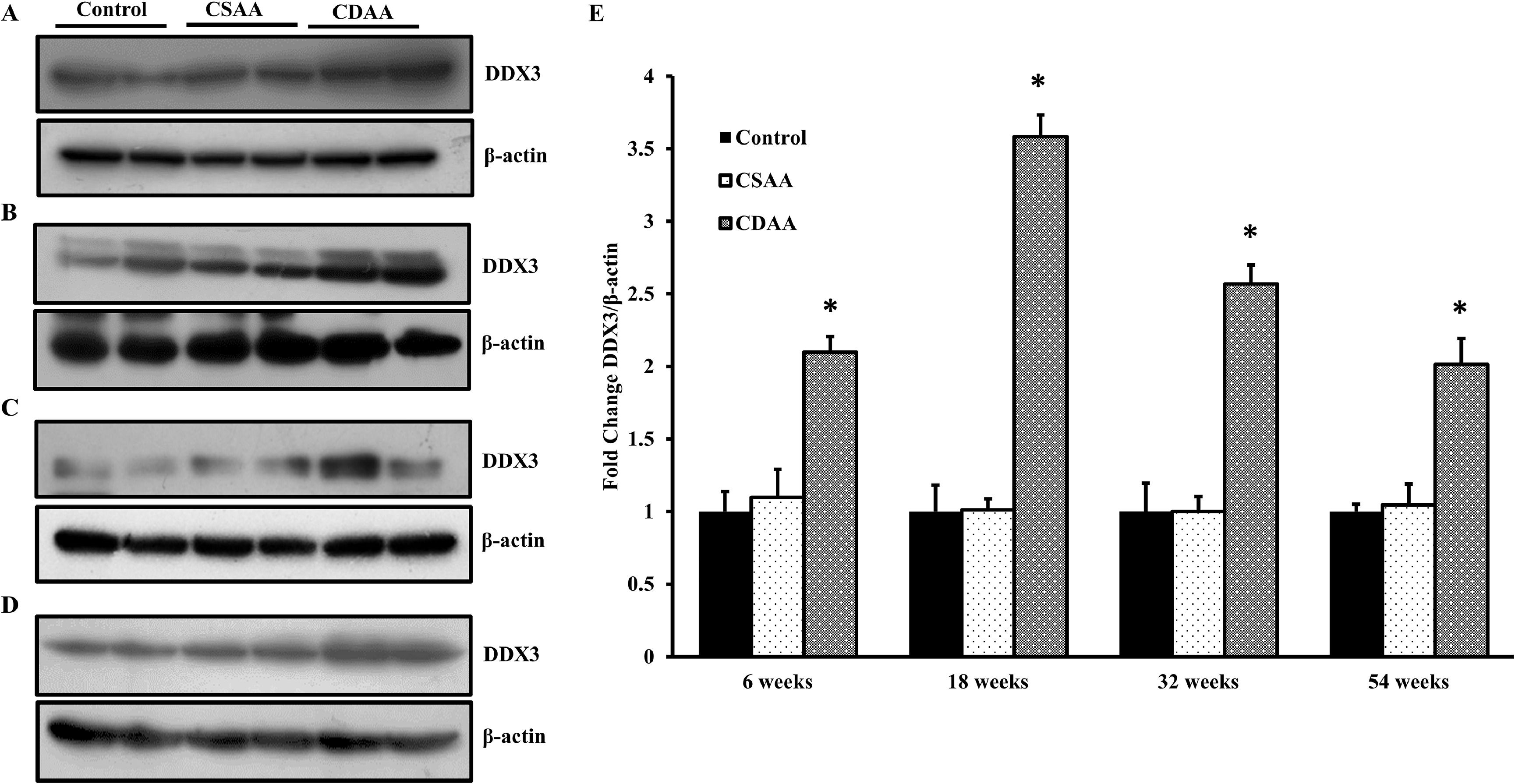
The in vitro results were validated using diet-induced mice model system. The C57BL/6 mice (6 mice per group) were fed with sham-operated control, CSAA, or CDAA diet for 6, 18, 32 and 54 weeks. The total protein or RNA was isolated from the liver after feeding the mice for various time points. Western blot analysis revealed that DDX3 expression was upregulated in the mice fed with CDAA diet compared to CSAA or sham-operated control mice in all the time points (Figs. 5A-5D). The band intensities were quantified and found that there was a 1.5-fold, 4.5-fold, 1.5-fold and 2-fold increase in the expression of DDX3 in 6, 18, 32 and 54 weeks CDAA fed mice, respectively (Fig. 5E).
, CSAA (control diet) and CDAA diet for 6 (A), 18 (B), 32 (C) and 54 (D) weeks. At the end of each time period, the total protein from the liver tissue was isolated and the expression of DDX3 and β-actin was determined. E. Densitometric analysis of the band intensities was determined by dividing the DDX3 expression with β-actin expression. The fold change was calculated taking control as 1, at each time point and the change in the expression of DDX3 was plotted. (n = 6; *p < 0.05).")
DDX3 upregulated in CDAA diet fed NAFLD mice. C57BL/6 mice were fed with chow diet (sham-operated control), CSAA (control diet) and CDAA diet for 6 (A), 18 (B), 32 (C) and 54 (D) weeks. At the end of each time period, the total protein from the liver tissue was isolated and the expression of DDX3 and β-actin was determined. E. Densitometric analysis of the band intensities was determined by dividing the DDX3 expression with β-actin expression. The fold change was calculated taking control as 1, at each time point and the change in the expression of DDX3 was plotted. (n = 6; *p < 0.05).
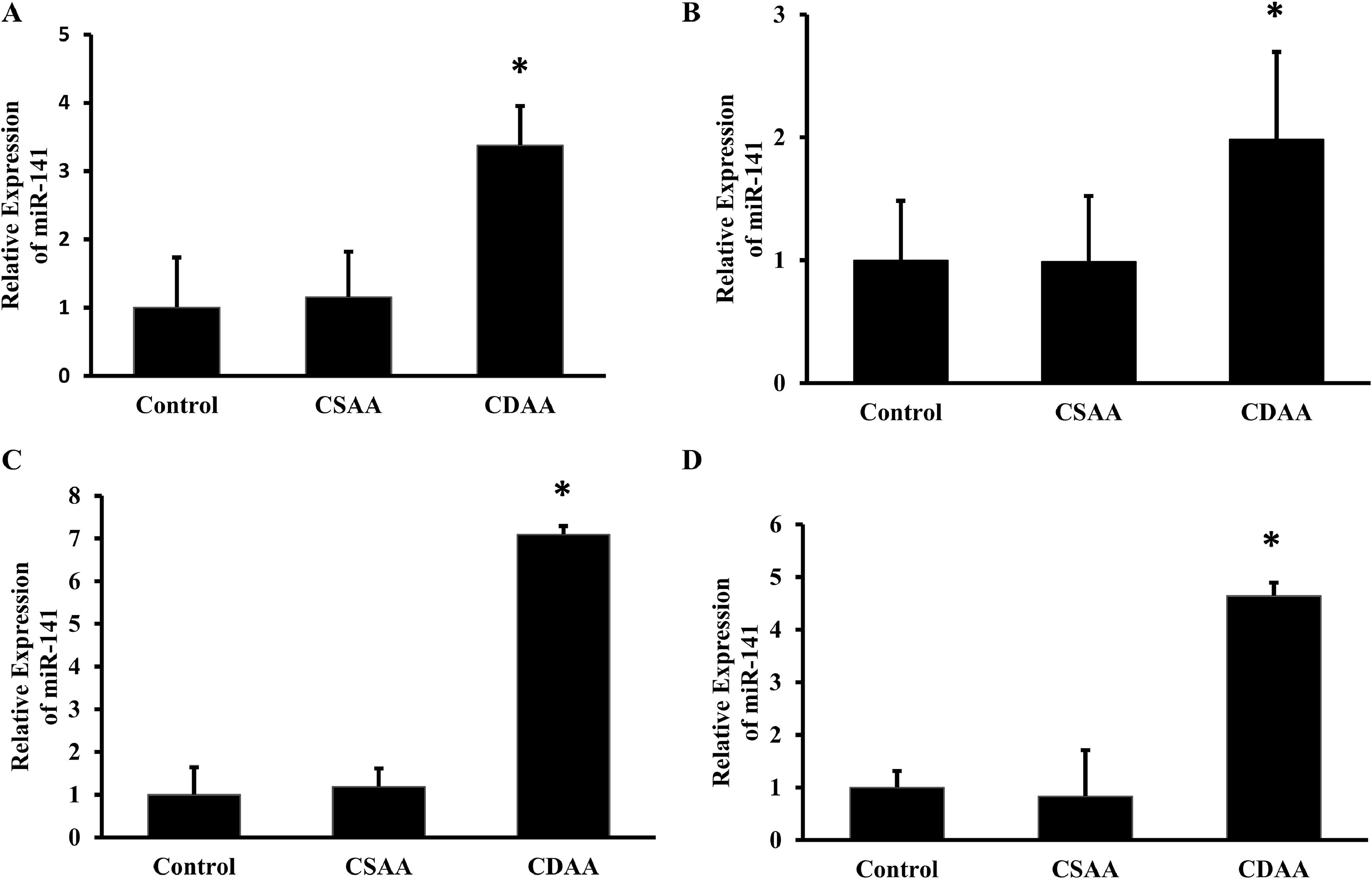
The total RNA isolated from the mice liver tissue was used for cDNA synthesis, followed by the real time PCR for miRNA-141. The results showed that there was a 3.3-fold, 2-fold, 7-fold, and 4.5-fold increase in the expression of miRNA-141 observed in the mice fed within 6, 18, 32 and 54 weeks CDAA fed mice, respectively (Fig. 6A-6D).
, 18 (B), 32 (C) and 54 (D) weeks and at the end of each time period, the total RNA was isolated from the liver tissue and Real time PCR was performed to determine the expression of miRNA-141 and 5S rRNA. The 2-ΔΔCT was calculated and then the relative expression was plotted by keeping control as 1-fold. The mean ± SD was calculated and the Student")
miRNA-141 expression was upregulated in CDAA fed mice. C57BL/6 mice was fed with sham-operated control, CSAA and CDAA diet for 6 (A), 18 (B), 32 (C) and 54 (D) weeks and at the end of each time period, the total RNA was isolated from the liver tissue and Real time PCR was performed to determine the expression of miRNA-141 and 5S rRNA. The 2-ΔΔCT was calculated and then the relative expression was plotted by keeping control as 1-fold. The mean ± SD was calculated and the Student's t-test was performed (n = 6; *p < 0.05).
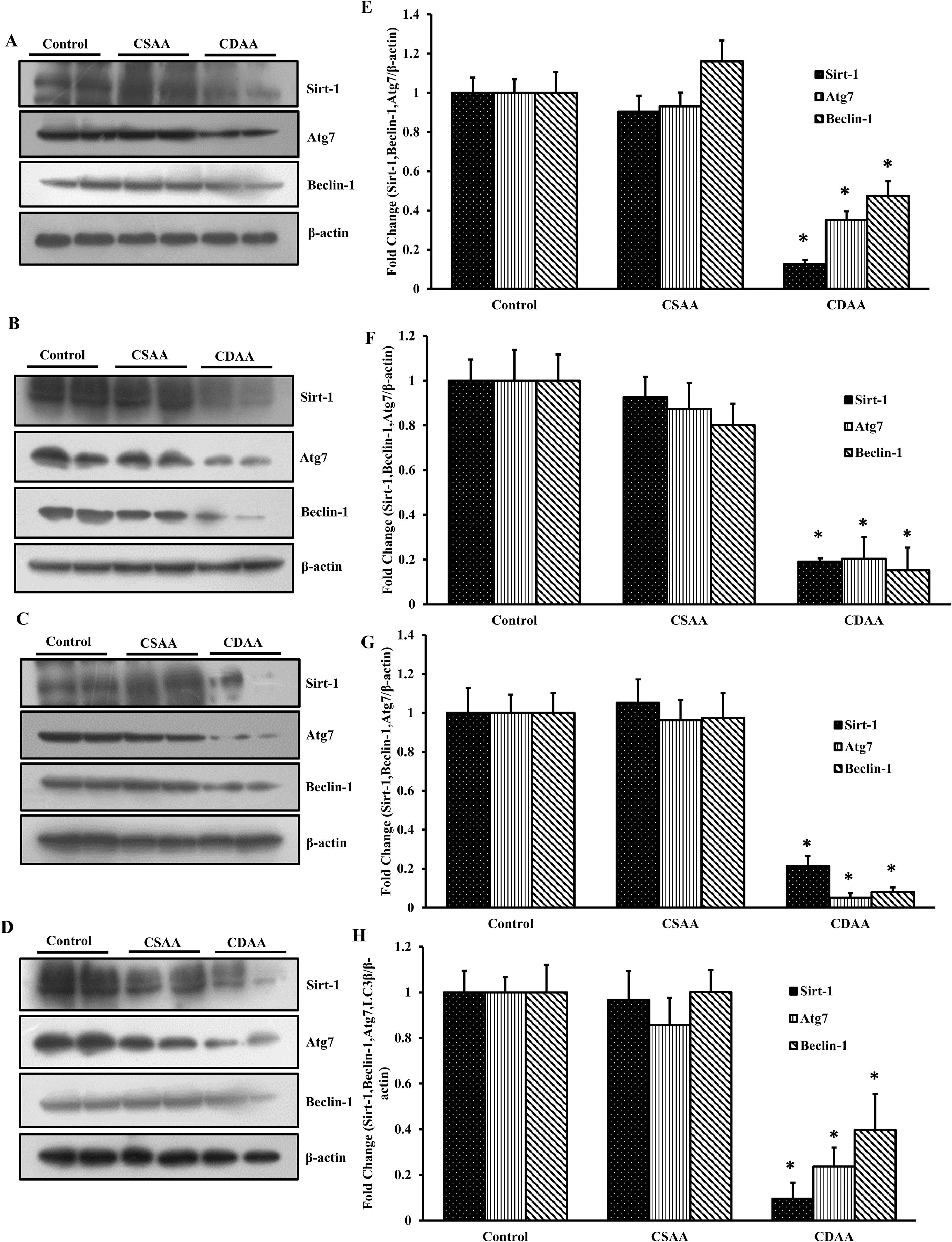
Western blots were performed from the total protein isolated from the mice liver from 6, 18, 32 and 54 weeks CDAA fed mice. The results showed that there was a significant decrease in the expression of autophagy marker proteins (Fig. 7A-7D). The band intensities showed that the expression of Sirt-1 was downregulated by 90 %, 80 %, 80 % and 90 % in 6, 18, 32 and 54 weeks CDAA fed mice, respectively (Fig. 7E-7H). Similarly, at least a 50 % to 70 % decrease in the expression of Atg7, beclin1 and LC3-II was observed in 6, 18, 32 and 54 weeks CDAA fed mice, respectively (Fig. 7E-7H).
, 18 (B), 32 (C) and 54 (D) weeks and at the end of each time period, the total protein was isolated from the liver tissue. Western blots were performed to determine the expression of Sirt-1, Atg7, beclin-1, and β-actin in the mice liver tissues. The band intensities was determined and determined by dividing the expression of gene of interest with β-actin expression. The fold change was calculated taking control as 1, at each time point and the change in the expression of the gene was plotted. E, F, G and H show the expression in fold change from the mice liver tissue isolated from 6, 18, 32 and 54 weeks after feeding, respectively (n = 6; *p < 0.05).")
Autophagy is decreased in CDAA fed mice. C57BL/6 mice was fed with sham-operated control, CSAA and CDAA diet for 6 (A), 18 (B), 32 (C) and 54 (D) weeks and at the end of each time period, the total protein was isolated from the liver tissue. Western blots were performed to determine the expression of Sirt-1, Atg7, beclin-1, and β-actin in the mice liver tissues. The band intensities was determined and determined by dividing the expression of gene of interest with β-actin expression. The fold change was calculated taking control as 1, at each time point and the change in the expression of the gene was plotted. E, F, G and H show the expression in fold change from the mice liver tissue isolated from 6, 18, 32 and 54 weeks after feeding, respectively (n = 6; *p < 0.05).
MASLD is one of the primary reasons for the development of hepatocellular carcinoma (HCC). MASLD progression to MASH and to HCC is a complex process involving the alteration of several signaling pathways. DDX3 performs many important functions in cellular processes and acts as a tumour suppressor in HCC [26]. It also acts as a double edged sword in various types of cancers [11]. So, as an oncogenic protein, DDX3 is highly overexpressed in breast cancer cell lines and promotes cell proliferation by inhibiting E-cadherin activity [7]. It also initiates the β-catenin signaling pathways to induce cell growth invasion of various cancers like colorectal cancer, gastric cancer and prostate cancer [27]. As a tumour suppressor protein, decreased DDX3 is negatively correlated with tumour formation, which results in poor progression of liver cancer in patients suffering from MASH, cirrhosis and liver related complications [17].
However, the exact role of DDX3 in MASLD is not fully understood. In this study, we established a crucial role of DDX3 in MASLD progression. DDX3 expression is significantly upregulated in Human HCC, which is triggered by exposure to diosgenin [28]. Similar to the previously published data, our study also showed that the expression of DDX3 was upregulated when incubated with FFAs. These results were confirmed in the diet induced MASLD mice model. DDX3 is associated with tumorigenesis in HCC patients, where DDX3 reduction is directly promoted tumour formation in hepatic cells [17]. To understand the mechanism by which DDX3 induces MASLD, the cells were incubated with the FFAs and the autophagy was determined and found to inhibit autophagy in hepatic cells. Previously, we had shown that DDX3 was inhibited by HBV, thereby inducing autophagy [15]. Similarly, others also found that DDX3 was involved in modulating autophagy. Autophagy is a highly conserved nutrients-recycling process where cells combat diverse stress including nutrient depletion. As a prime novel regulator, DDX3 binds with 5ʹ-UTR of ATG1 to resolve its secondary structure and eventually modulates autophagy during nitrogen starvation relative to amino acids starvation [24]. This study also demonstrated that DDX3 positively regulates ULK1 expression at post transcriptional level to promote autophagy in mammalian cells and knockdown of DDX3 leads to reduction of autophagy. Thus, DDX3 has a direct interplay between autophagy and its related pathways.
Previously, several studies have shown that DDX3 levels were positively correlated with the expression of miRNAs like miR-200b, miR-200c, miR-122, miR-145 [29]. As a RNA binding protein, DDX3 interacts with Drosha/DGCR8 complex (miRNA microprocessor), promotes pri-miRNA processing activity and finally elevates mature miRNA expression level [30]. Previously, it was shown that DDX3 could upregulate a subset of miRNAs, including miRNA-141, which played important role in the cancer progression [30]. These authors showed that though DDX3 interacted with Drosha/DGCR8 complex, the DDX3’s KO/OE strategies seemed to affect a small part of miRNA expression levels, including miRNA-141. miRNA-141 was shown to target Sirt-1and inhibit autophagy to reduce HBV replication [31]. Hence, we determined the expression of miRNA-141 in FFA incubated cells and found that there was an increase of miRNA-141 in hepatic cells. To study whether this increase is due to the DDX3, the cells were transfected with DDX3 expression plasmid and found a positive correlation between DDX3 expression and miRNA-141 expression. Further, inhibition of DDX3 using siRNAs resulted in decreased miRNA-141expression, confirming that DDX3 is directly involved in regulating miRNA-141 expression. Using bioinformatics approaches, such as Targetscan, it was found Sirt-1 was a target for miRNA-141. Hence, miRNA-141 overexpression experiments showed that there was a significant decrease in the expression of Sirt-1. Similar results were observed when DDX3 was overexpressed in HepG2 cells, whereas the expression of Sirt-1 was increased when DDX3 was inhibited using siRNAs to DDX3. This clearly established that FFAs induced the DDX3, which in turn upregulated the expression of miRNA-141. This was further confirmed by the expression of autophagy marker protein; expression was inhibited when DDX3 was overexpressed and autophagy was induced when DDX3 was inhibited. Previously, two dead box RNA helicase family members, DDX5 and DDX10, have been reported to modulate autophagy [32,33].
Studies showed that miR-141 inhibits Sirt-1 expression and promotes cells proliferation and reduces apoptosis in colon cancer cells [34]. Zhou et al. recently demonstrated that miR-141 targeted Sirt-1 and downregulated its expression to inhibit osteogenic differentiation of bone marrow mesenchymal stem cells (BMSCs) [35]. All these studies support our experiments that show that miR-141 has a direct positive correlation with Sirt-1 expression.
5ConclusionsFor the first time, we showed that DDX3 inhibits autophagy via miRNA-141 in MASLD. To confirm these in vitro findings, the CDAA diet-induced mice model system was used. CDAA diet has high sucrose (40 %) and fat (10 %), and the proteins are replaced with an equivalent and corresponding mixture of L-amino acids [36]. In hepatocytes, this diet inhibits fatty acid oxidation while it increases lipid synthesis, oxidative stress, and inflammation, resulting in liver fibrosis [37], but the development of histological changes requires a longer time frame [36]. Hence, the mice were fed up to 54-week time period. Mice on the CDAA diet neither display hepatic insulin resistance [38] nor gain weight. The establishment and the confirmation of the MASLD mice model using histological analysis of liver tissue measurement of TG and HbA1c were performed and found that the disease progression was increasing with the feeding interval of CDAA diet, and all these results had already been published by our group [25]. As MASLD pathogenesis progressed, the expression of both DDX3 and miRNA-141 increased. Autophagy was also inhibited in mice fed with CDAA diets. Autophagy is a self-eating process to maintain cellular quality control and acts as a source of energy during starvation, which also acts as a protective mechanism for the progression of cancers[39,40]. Autophagy also degrades lipid droplets through lipophagy in the liver as well as various cell types in yeast, worms, and fungal structures [41]. Thus, considering the importance of autophagy in cells, we developed a novel therapeutic target of MASLD progression towards MASH and HCC by targeting Sirt-1/autophagy axis through miR-141. Our findings identified a novel mechanistic framework in which the upregulation of DDX3 expression in MASLD enhanced miR-141 levels, which in turn downregulated Sirt-1 and consequently, a reduction in autophagic activity was observed.
Author contributionsThe corresponding author planned the study and corrected the manuscript. Md. Musa Hossain and the corresponding author designed all the experiments. Md. Musa Hossain performed all the experiments, analysed the data and wrote the manuscript. Amit K. Mishra helped in designing the experiments and performed some of the transfection and western blot experiments and also helped in preparing manuscripts. Ajay K. Yadav and Md. Musa Hossain established NAFLD mice model system & Sata Teja Naveen, Md. Ismail, Amrendra K. Sah assisted in mice tissue collection, few cell culture and Western blot experiments. Arnab Banik and Gopal Sharma performed some of the western blots experiments related to autophagy. All the authors have read the manuscript and declared no potential conflict of interest regarding this research.



