



Treatment of chronic hepatitis C with type I interferons and ribavirin can be associated with exacerbation of hepatitis and sometimes liver decompensation. We report two patients with chronic hepatitis C virus infection who experienced a severe increase of bilirubin levels of up to 17 times upper the limit of normal value in the absence of deterioration of hepatic function during therapy with pegylated-interferon and ribavirin. A genetic disposition for Gilbert’s syndrome explained the adverse events and permitted a continuation of therapy leading to a sustained clearance of chronic hepatitis C infection. Since one patient jaundiced already during a lead-in treatment period with ribavirin monotherapy we suggest that hyperbilirubinaemia during combination therapy is primarily caused by ribavirin rather than by effects of interferon alpha on UDP-glucuronosyltransferase activities. Of note, both patients recovered from their initial unconjugated hyperbilirubinemia despite continuation of ribavirin therapy, which indicates that compensatory mechanisms leading to a normalization of UGT1A1 activity are likely.
The standard therapy for patients with chronic hepatitis C virus infection (HCV) consists of pegylated-interferon (PEG-IFN) plus ribavirin. Patients with HCV-genotype 2 or 3 infection are treated for 16-24 weeks, whereas patients with HCV-genotype 1 require 24-72 weeks of therapy, leading to sustained virological response rates (SVR) of 50% in genotype 1 and 80%-90% in genotype 2 or 3 infection.1-4 Treatment can be associated with significant side effects such as the induction of autoimmune disease, flu-like symptoms and psychiatric symptoms such as depression and agitation.5 The exacerbation of hepatitis activity and even liver decompensation are serious adverse events, which can occur during interferon treatment.6 Increased activity of aminotransferases in serum and particularly ALT flares during Interferon-based therapies have been described to occur in a considerable proportion of patients.5,7,8 In clinical practice the challenge is to determine the precise aetiology of elevated liver specific parameters in order to decide, whether to continue treatment and possibly achieve remission, or to stop therapy avoiding serious deterioration of the patient’s condition but also decreasing the chance of a cure. This is regularly the case when marked elevations of aminotransferase activities or serum bilirubin concentrations are observed under antiviral therapy.
We report two patients with chronic hepatitis C virus infection who experienced a severe increase of bilirubin levels as early as nine and 13 days after the initiation of therapy, in whom a genetic disposition explained the adverse effects and permitted a continuation of therapy leading to a sustained clearance of HCV infection.
Case 1A 22-year-old male patient with chronic hepatitis C virus infection, genotype 2 was referred to our outpatient clinic for evaluation and initiation of antiviral treatment in October 2003.
The acquisition mode of HCV infection was unclear. The history revealed intermittent hyperbilirubinemia since childhood which had not been further diagnostically worked up. Prior to treatment blood count and prothrombin time were normal, ALT and bilirubin levels were increased to 82 U/l (normal range < 45 U/l) and 94 pmol/l (normal range < 17 pmol/l), respectively. HCV-RNA viremia was greater than 500,000 I.E./mL (Cobas Amplicor Roche Diagnostics, Germany).
The patient was treated within the REDD 2/3 study of the German Network of competence on viral hepatitis (Hep-Net)9 and was randomized to receive weight-adjusted antiviral combination treatment with PEG-IFN-alfa 2b 10 μg/oiw and ribavirin 1,000 mg/d.
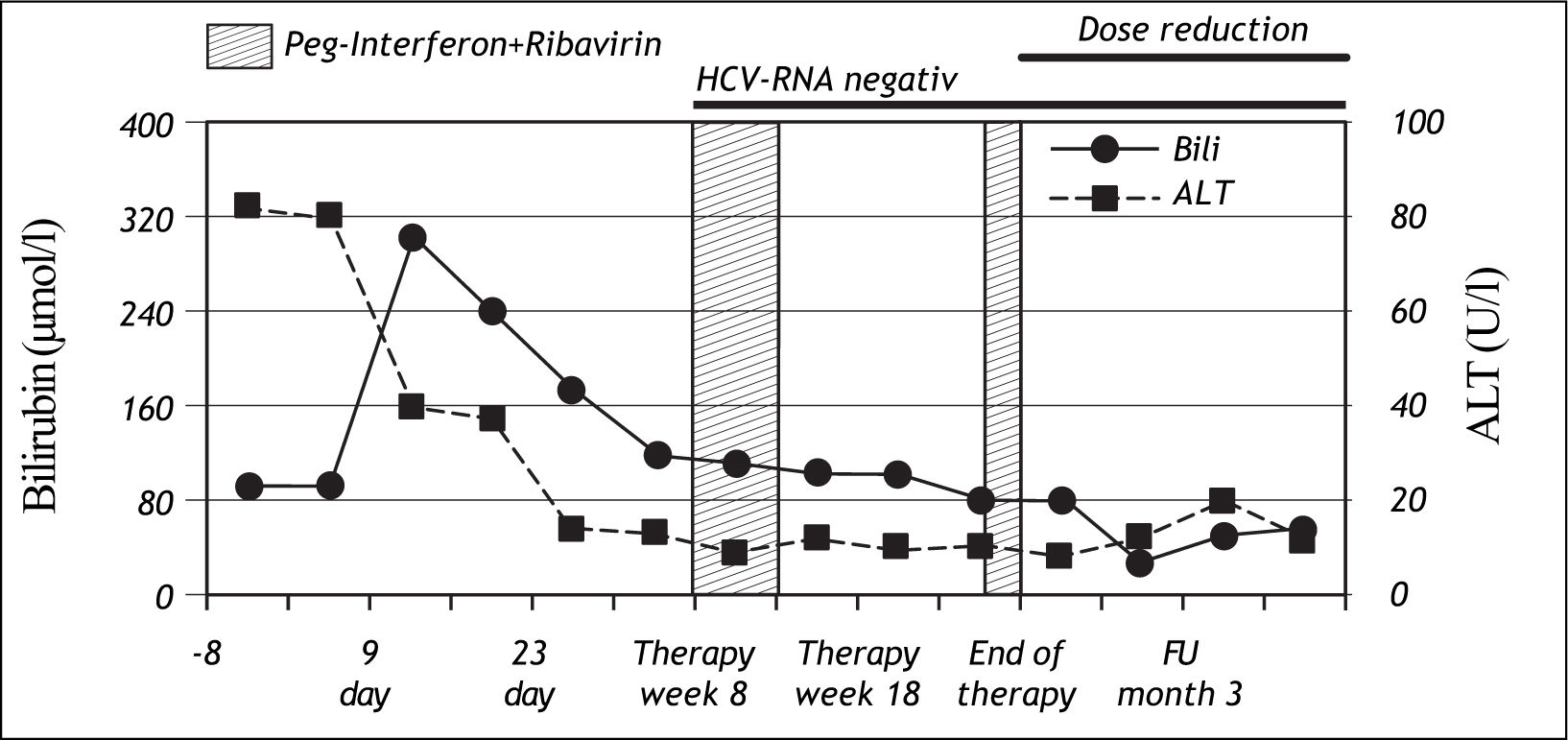
Nine days after the first injection of PEG-IFN alfa-2b and administration of ribavirin, the patient developed severe jaundice without pain, pruritus or any general symptoms. At this time, ALT levels had normalized, but total bilirubin was increased to 303 μmol/l. (normal range: < 17 μmol/l). A sub analysis showed that conjugated bilirubin was < 2 μmol/ l (normal range: < 2 μmol/l), and unconjugated bilirubin was 290 μmol/l (normal range: < 17 μmol/l) (Figure 1), lactate dehydrogenase and haptoglobin levels were normal. Genotyping for Gilbert’s syndrome was performed by DNA sequencing showing homozygosity for a TA insertion into the UDP-glucuronosyltransferase 1A1 (UGT1A1) promoter region (UGT1A1*28), representing the most common variant associated with Gilbert’s syndrome in Caucasians.

Based on the hypothesis of Gilbert’s associated unconjugated hyperbilirubinemia in the absence of inflammatory exacerbation or a deterioration of hepatic function antiviral treatment was continued.
During the course of therapy bilirubin levels decreased, baseline values were reached at week 22. Unconjugated bilirubin was found to be elevated throughout the course of therapy while conjugated bilirubin was normal.
In addition to hyperbilirubinaemia, the patient suffered from common interferon-related side effects such as flu-like symptoms and mild arthralgia. Moreover, interferon induced leukopenia led to a dose reduction between therapy week 8-13 and therapy week 22-24. Dose reduction of ribavirin was not necessary during the whole period of therapy. HCV-RNA was negative for the first time at week 8 and remained negative for 2 years beyond the end of therapy when the patient was seen the last time at our department.
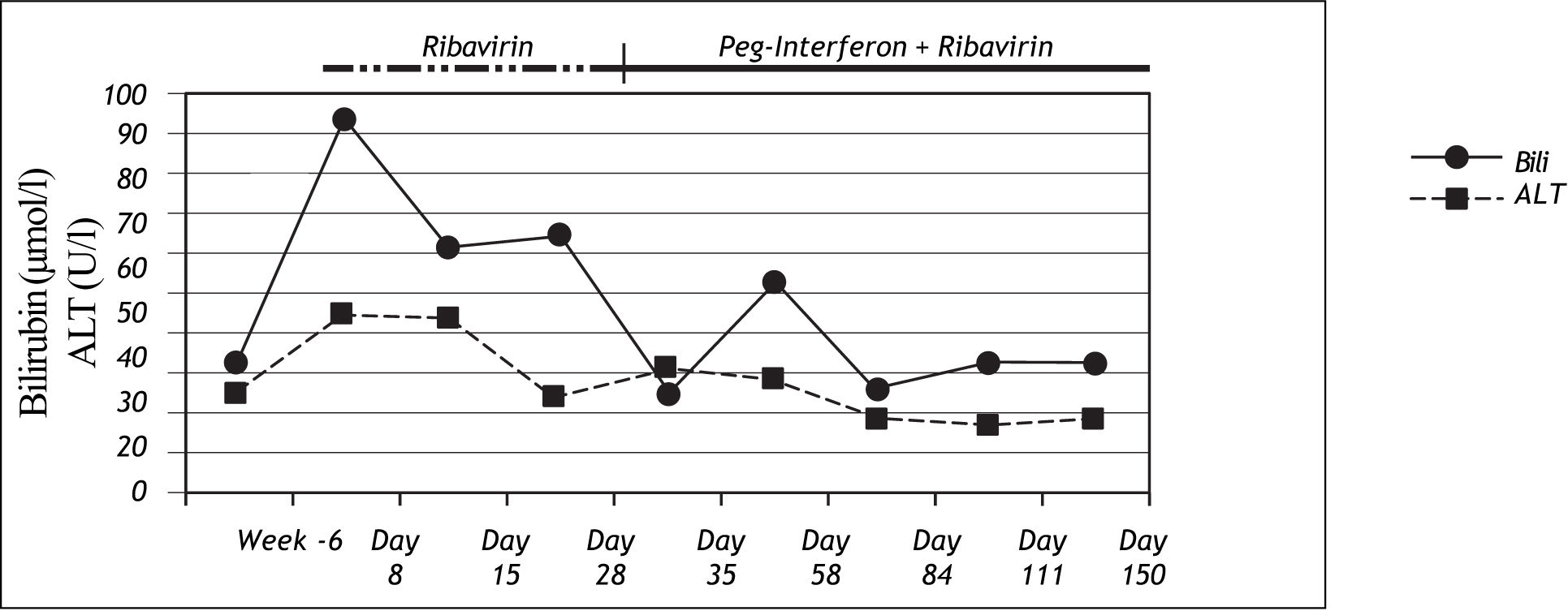
Case 2A female patient with chronic hepatitis C virus infection, genotype 1b, 62 years old, began antiviral therapy with ribavirin 1,000 mg/d per day in September 2006. The acquisition mode was postpartal anti-D-prophylaxis in the year 1979 as recently described.10 HCV-RNA was detected for the first time in 1994. She had no history of intermittent hyperbiliru-binemia. She first received ribavirin monotherapy since the patient had thrombocytopenia and the known effect of ribavirin to increase platelet levels11 was anticipated to allow the usage of optimal doses for subsequent interferon alfa treatment. Eight days after the initiation of ribavirin treatment, the patient showed severe jaundice without additional symptoms, abdominal pains or pyrexia. At this time her ALT activities were normal, and bilirubin had increased to 94μmol/l (normal range: <17μmol/l). Genotyping revealed the homozygous for UGT1A1*28 variant. Ribavirin was reduced to 400mg/d for 3 weeks due to anaemia and could be increased again to 800mg/d during the course of treatment. Peg-Interferon alfa-2b treatment was started 4 weeks after the start of ribavirin and could only be administered in a reduced dose of 50 μg per week due to adverse events. Antiviral treatment was stopped after 16 weeks of therapy due to virological non-response. Bilirubin levels decreased to normal values rapidly again after stopping the antiviral treatment (Figure 2).
Discussion
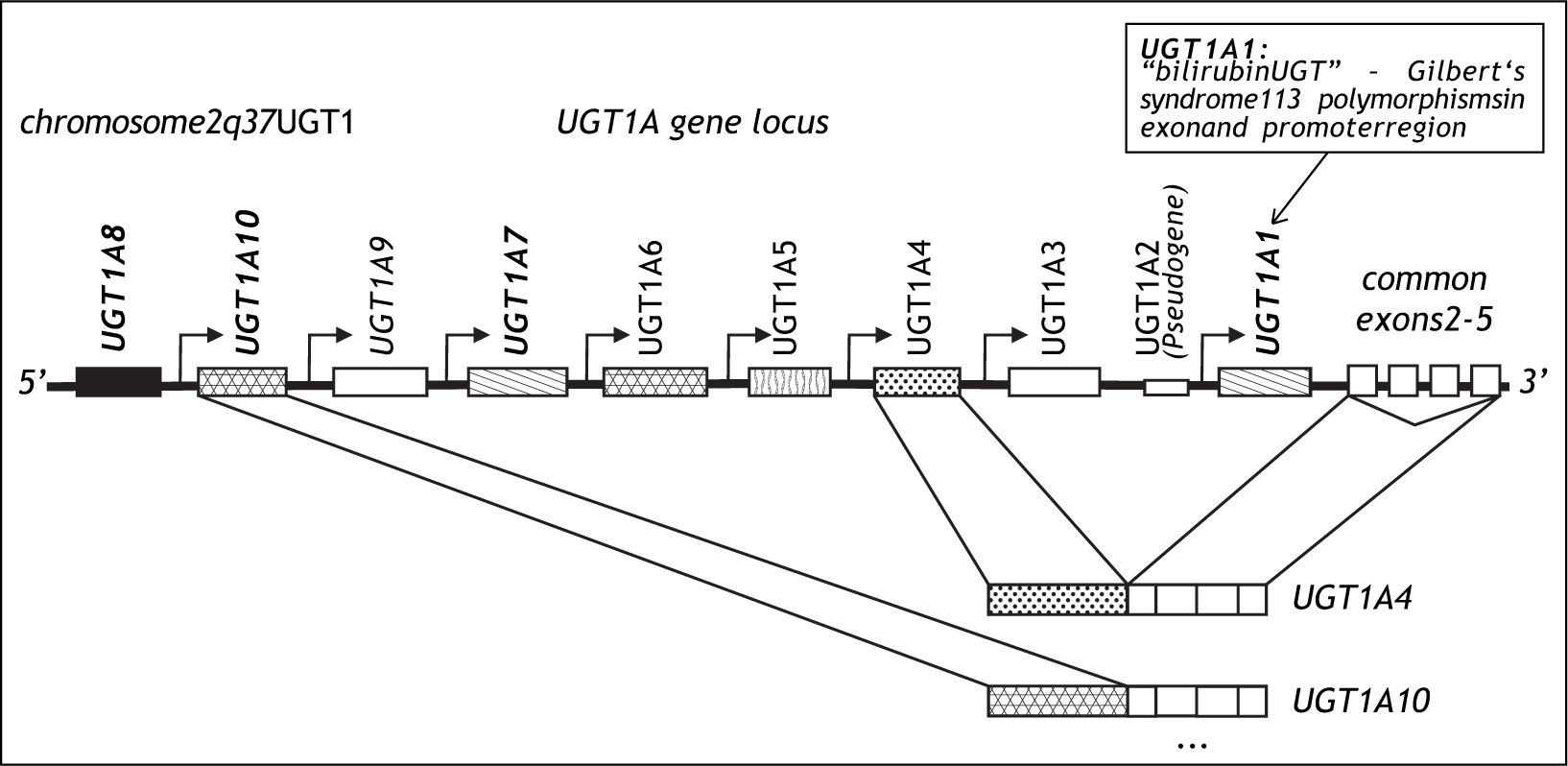
Gilbert’s syndrome is one of the most frequent genetic variants of metabolism in Caucasians.12 It is characterized by a 70% reduction of bilirubin glucuronidation caused by a TA insertion into the TATA box of the UflP-glucuronosyltransferase (UGT)1A1 gene designated UGT1A1*28.13,14 UGT1A1 was cloned in 1991 and is a member of the UGT1A gene family encoded on chromosome 2,15 which leads to the tissue specific transcription of 9 UGT1A genes leading to 9 UGT1A protein isoforms16 (Figure 3). UGT1A1 is expressed primarily in the liver but also in small intestine.17 It is the only efficient physiological enzyme capable of generating water soluble bilirubin glucuronides, which can be eliminated from the body via urine and bile,18 and its functional variation therefore leads to the clinical phenotype of mild non-haemolytic hyperbilirubinemia. A complete loss of bilirubin glucuronidation is fatal and is the cause of Crigler-Najjar type 1 disease.19 The promoter polymorphisms UGT1A1*28 was associated with Gilbert’s syndrome in 1995.13,14 With an allelic frequency of 0.4 homozygous carriers of UGT1A1*28 occur in 16%, although the clinical phenotype is only observed in approximately 10% of the population. Gilbert’s syndrome is characterized by episodic increases of unconjugated serum bilirubin rarely exceeding 150 μM and usually in response to fasting or stress, and this condition does not lead to liver inflammation, fibrosis or progressive liver damage.20 In an unchallenged individual, Gilbert’s syndrome is therefore most often unapparent. However, recent studies have reported pharmacogenetic risks associated with the presence of the UGT1A1*28 genotype. The risk of severe irinotecan drug toxicity has been associated with UGT1A1*2821 as well as with a combination of UGT1A1 and UGT1A7 polymorphisms.22 In addition protease inhibitor associated jaundice has been linked to a haplotype of UGT1A1*28 in combination with variants of the UGT1A3 and UGT1A7 genes.23 These data suggest, that this benign condition is in fact a risk factor for drug associated toxicity and jaundice.12

Our example of two patients receiving antiviral therapy for chronic HCV infection adds another example to these reports, which potentially affects a considerable number of patients treated for HCV infection. In the first patient, a marked increase of total bilirubin levels-almost exclusively unconjugated after sub analysis-was observed within only 9 days after the initiation of subcutaneous PEG-IFN-alfa 2b and oral ribavirin. This coincidence would suggest a direct effect of either interferon-alpha or ribavirin, or both, on the activity of UGT1A1. It could be hypothesized that pro-inflammatory activity of interferon may mimic stress such as during fasting or infection, which is capable of elevating bilirubin in individuals with Gilbert’s syndrome. However, major trials of interferon monotherapy have not reported an increase of bilirubin although it can be expected that approximately 10% of Caucasian study populations are carriers of the homozygous UGT1A1*28 genotype. A second hypothesis would concern ribavirin, which is a potent cause of haemolytic anaemia, and may have increased the amount of free bilirubin, thereby generating an excess of UGT1A1 substrate. This in turn would not be sufficiently glucuronidated by UGT1A1 thus leading to high level unconjugated hyperbilirubinemia, which in our patient clearly exceeded levels expected in an episode ofjaundice of an individual with Gilbert’s syndrome. It is interesting to note, that the history of patient 1 was positive for occasional episodes of scleral jaundice, which had previously not been further analysed and did not bother the patient to the extent to be mentioned to his physicians prior to HCV treatment. The treatment course of the second patient provides arguments for an effect of ribavirin. In this patient with Gilbert’s syndrome ribavirin treatment was initiated in the absence of interferon leading to the same rise in bilirubin within 13 days of treatment initiation. This observation indicates that HCV treatment associated jaundice does not require interferon. The mechanisms of this effect may include increased haemolysis, direct inhibition of UGT function,24,25 or an effect on the transcriptional regulation of the UGT1A1 gene,26 which is already reduced by 70%. Of note, both patients recovered from their initial unconjugated hyperbilirubinemia despite continuation of ribavirin therapy, which indicates that compensatory mechanisms leading to a normalization of UGT1A1 activity are likely.27 The fact that 16% of Caucasians carry this variant and marked jaundice in HCV therapy only rarely occurs also indicates that additional variants, which may include transporter proteins, may play a permissive role. From a clinical point of view the observation in these two patients is of high practical consequence. Elevations of aminotransferases and/or cholestatic markers are likely to lead to the decision to discontinue antiviral therapy eliminating the chances of a cure. However, unconjugated hyperbilirubinemia based on lower UGT1A1 activity is not life threatening. It only leads to a clinical symptom without further relevant morbidity, and cannot be viewed as a contraindication for therapy. In this situation genotyping and establishment of the Gilbert’s genotype provided a diagnostic basis to continue therapy eventually permitting the realization of a sustained virological response.
In conclusion, IFN-alfa and ribavirin therapy can induce severe unconjugated hyperbilirubinaemia in patients with Gilbert’s syndrome, which is linked to ribavirin. Continuation of therapy is possible, safe and effective in these individuals. Hyperbilirubinemia was observed to improve during the course of continued therapy. In unexpected hyperbilirubinemia during the initial phase of antiviral therapy determination of unconjugated bilirubin and genotyping for UGT1A1*28 is suggested for the decision to continue therapy.
Abbreviations- •
HCV: Chronic hepatitis C virus.
- •
PEG-IFN: Pegylated interferon.
- •
SVR: Sustained virological response rates



