




Recent studies have suggested an association between H. pylori and metabolic dysfunction associated steatotic liver disease (MASLD). We aim to evaluate the association of H. pylori virulence genes with non-invasive markers of liver injury and fibrosis in MASLD subjects.
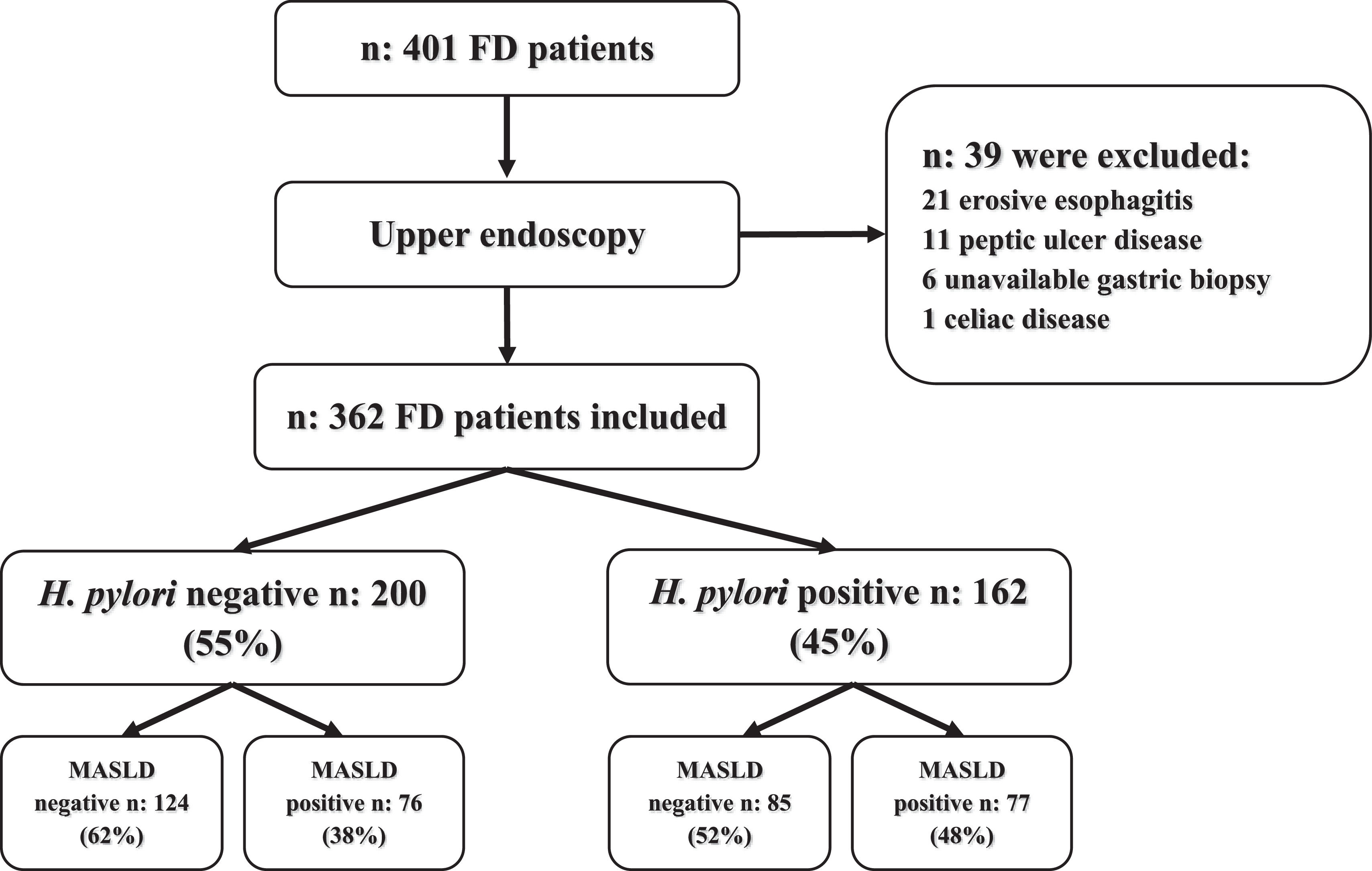
Patients and MethodsA total of 362 dyspeptic patients who underwent gastroscopy were selected. Biochemical, clinical parameters, ultrasound, FIB-4 score, liver stiffness measurement (LSM) by vibration-controlled transient elastography (VCTE), gastric biopsies, and H. pylori virulence genes (cagA, vacA) were evaluated.
ResultsA cohort comprised of 61 % women and 39 % men with a median age of 52 (40–60) years. MASLD was observed in 42 %, and H. pylori-positive in 45 %. No differences were observed regarding H. pylori status at co-morbid metabolic conditions. In MASLD cohort, H. pylori-positive was associated with higher AST, ALT, FIB-4 and LSM. Indeed, carriers of cagA/vacA-s1/m1-positive allelic combination were associated with higher AST, ALT, FIB-4 and LSM but not cagA/vacA-s1/m1-negative. The OR for high-risk of significant/advanced- fibrosis by VCTE (≥8 kPa) with H. pylori-positive was 2.56 (95 % CI, 1.2–5.75) and for cagA/vacA-s1/-m1-positive allelic carriers was 4.01 (95 % CI, 1.38–11.56), but non-significant association in cagA/vacA-s1/-m1-negative. After adjusting for age, gender, diabetes, BMI and hypertension the OR for VCTE ≥8 kPa with H. pylori-positive was 2.43 (95 % CI, 1.88–12.44), and cagA/vacA-s1/m1-positive allelic carriers was 4.06 (95 % CI, 1.22–14.49).
ConclusionsIn our cohort of functional dyspepsia (FD) patients with MASLD, H. pylori was associated with non-invasive markers of liver injury and fibrosis. Carriers of cagA/vacA-s1/m1-positive allelic combination showed an independent risk of significant/advanced fibrosis by VCTE.
Metabolic-dysfunction-associated steatotic liver disease (MASLD), previously known as Nonalcoholic Fatty Liver Disease (NAFLD) is currently the most common chronic liver disease [1,2]. The global prevalence of MASLD has increased from 25 % in 1990–2006 to 38 % in 2016–2019 [3]. Its prevalence is rising worldwide in parallel with increases in obesity and metabolic comorbid disease (insulin resistance, diabetes mellitus and obesity) [4,5]. MASLD encompasses a histological spectrum of disease characterized by macrovesicular hepatocyte steatosis that may be accompanied by mild inflammation, and metabolic dysfunction associated steatohepatitis (MASH), the more active form of MASLD characterized by the presence of hepatic steatosis, inflammation, and hepatocyte ballooning, is emerging as one of the leading causes of cirrhosis, cirrhotic complications, hepatocellular carcinoma (HCC), liver transplantation and liver-related death [1]. MASLD is a complex disorder that is influenced by multiple mechanisms, including genetic, environmental, and metabolic factors [6]. The pathogenesis, particularly the drivers of progression in MASLD are incompletely understood [7].
Helicobacter pylori (H. pylori) is a gram-negative, microaerophilic bacteria that colonizes gastric mucosal epithelium [8]. Similarly to the high prevalence of MASLD, H. pylori has highly global prevalence, affects nearly 50 % the world's population with a higher prevalence in developing countries and is a key constituent of the human microbiome [8]. Clinical manifestations of H. pylori infection include peptic ulcer disease, gastric mucosa-associated lymphoid tissue lymphoma, and non-cardia gastric adenocarcinoma [9]. H. pylori is also the main infectious-related ethological cause of functional dyspepsia (FD) [10]. H. pylori infection not only affects gastric mucosa but is also linked to a number of extra-gastric diseases, indicating that H. pylori may cause disease far from the primary site of infection by a different pathogenic process [11]. Notably, recent attention has focused on whether H. pylori infection contributes to metabolic disorders including MASLD [12,13]. However, the existing literature on the association between H. pylori infection and MASLD is conflicting, with some studies reporting a positive association while others finding no significant relationship [14,15]. H. pylori is thought to contribute to the pathogenesis of MASLD by increasing insulin resistance, stimulating the release of proinflammatory cytokines, and increasing intestinal permeability [16,17]. The interaction between H. pylori virulence genes and human genetic polymorphisms in the pro- and anti-inflammatory cascade appear to play a role in the host's susceptibility to H. pylori gastritis and duodenitis [18,19]. Notably, H. pylori virulence genes, mainly cytotoxin-associated gene A (cagA) pathogenicity island (PAI) and vacuolating cytotoxin A (vacA), determine H. pylori pathogenicity and pro-inflammatory response [9,20]. Moreover, H. pylori internalization and injury in hepatocytes is also related on cagA and vacA status [21,22]. In this regard, previous data on chronic hepatitis C showed an association between H. pylori cagA status in the progression to cirrhosis and hepatocellular carcinoma [23]. It is noteworthy, that H. pylori infection per se is linked to gut dysbiosis including alterations in bacterial diversity and abundance that may influence metabolic derangements [17]. Viewing aforementioned data, we can consider that H. pylori virulence genes may contribute to MASLD severity. Thus, our aim was to evaluate the association of H. pylori virulence genes with non-invasive markers of severity in MASLD subjects. To address this subject, we examined the role H. pylori gastric infection, cagA and vacA virulence genes status in MASLD diagnosis, non-invasive markers of liver injury and fibrosis using a FD population who underwent upper endoscopy.
2Patients and Methods2.1PatientsWe consecutively recruited patients newly diagnosed with FD who were scheduled to undergo gastroscopy from January 2018 to December 2021 at IOT Medical Center, Simes Medical Institute (Posadas, Province of Misiones) and University Hospital San Juan Bautista (Santo Tomé, Province of Corrientes), Argentina.
The criteria for inclusion were: (1) age between 18 and 70 years, (2) symptoms meeting Rome-III criteria. The criteria for exclusion before gastroscopy were: (1) progressive, severe diseases requiring active medical management (e.g. advanced congestive heart failure, uncontrolled diabetes, decompensated cirrhosis, end-stage renal failure, neurological disease, advanced cancer, or psychiatric disorder), (2) those with known causes of chronic liver diseases and significant alcohol consumption (defined as ≥ 140 g/week for women and ≥ 210 g/week for men), (3) autoimmune medical conditions (inflammatory bowel disease, celiac disease, vasculitis, connective tissue disease), (4) atopic disease such as food allergies (milk, eggs, peanuts, tree nuts, fish, shellfish, fruit, and vegetables), asthma, allergic rhinitis or drug reaction, and eczema, (5) patients who had taken steatogenic medications (corticosteroids, tamoxifen, amiodarone, methotrexate), (6) patients who had taken antibiotic within the past 3 weeks and (7) history of previous H. pylori infection and gastric or bariatric surgery. Patients receiving proton pump inhibitors (PPI), H2-blockers or non-steroidal anti-inflammatory drugs (NSAID) were advised to suspend them 14 days before endoscopy. The criteria for exclusion after upper endoscopy were: (1) evidence of active peptic ulcer disease or gastro-esophageal erosive esophagitis, (2) evidence of malignant gastric disease, (3) signs of celiac disease, and (4) not available gastric biopsies.
2.2Clinical variablesClinical co-morbid conditions and anthropometric variables were obtained by a pre-endoscopy interview, data regarding age, sex, body mass index (BMI), alcohol consumption, smoking habits, medical history, including presence of hypertension [24], pre-diabetes/type-2 diabetes [25], surgery, malignancy and chronic liver disease were collected. Medical history also involved the regular utilization of NSAIDs, PPI, H-2 blockers, anti-hypertensive medications and anti-diabetic medications. The following laboratory parameters were analyzed in this study: aspartate aminotransferase (AST), alanine aminotransferase (ALT), gamma-glutamyl transferase (GGT), alkaline phosphatase (ALP), platelets and viral hepatitis serology (HBsAg, Anti-HBc and Anti-HCV). AST and ALT cut-off of upper limit of normal were defined as <40 UI/mL [26]. The FIB-4 index was calculated using the formula: FIB-4 = Age (years) × AST (IU/L)/[PLT(109/L) × √ALT (IU/L)] [27]. Previously published FIB-4 cut-off were used to rule-out (<1.3) advanced fibrosis (AF) [28]. Cut off of FIB-4 ≥ 1.3 is recommended to identify patients with MASLD as intermediate-high risk of AF requiring referral to the specialist liver clinic [28].
2.3FD and MASLD definitionFD patients satisfied the Rome-III criteria for the past three months with symptom onset at least six months before diagnosis [29]. FD was divided into 2 subtypes depending on the symptoms: Epigastric pain syndrome (EPS) is associated with epigastric pain or epigastric soreness, and postprandial distress syndrome (PDS) is associated with early satiety or postprandial fullness. Those who meet both criteria were classified as EPS/PDS overlap syndrome. MASLD was defined according to the multi-society consensus statement on new fatty liver disease nomenclature [2], the diagnosis of MASLD required the following: (1) hepatic steatosis detected by ultrasonography; (2) no significant alcohol consumption (defined as <140 g/week for women and <210 g/d for men); (3) the presence of one cardiometabolic risk factor and (4) no other discernible cause of steatosis.
2.4Vibration controlled transient elastographyVibration controlled transient elastography (VCTE) was performed by FibroScan-402 (Echosens, Paris, France) medical device using the M or XL probe as appropriate. Liver stiffness measurement (LSM) was assessed after a diagnosis of MASLD by ultrasound, by one expert operator. Measurements were performed on the right lobe of the liver through intercostal spaces guided by ultrasonography with the patient lying in dorsal decubitus with the right arm in abduction. LSM was expressed in kilopascal (kPa) and calculated as the median value of ten successful acquisitions, defined by a success rate of at least 60 %, and by an interquartile range lower than 30 % [30]. Previously published LSM cut-offs were used to rule-out Fibrosis stage-2 or greater stage (significant fibrosis, SF) ≥5.8 kPa, and to rule-out Fibrosis stage-3 or greater stage (advanced fibrosis, AF) ≥8 kPa respectively [31,32,33].
2.5EndoscopyAll recruited participants underwent gastroscopy performed by experienced endoscopists. Biopsy specimens were collected from the lesser curvature of the gastric body (two biopsies) and lesser curvature of the gastric antrum (two biopsies).
2.6Histopathologic analysisBiopsies were fixed in 10 % formalin and processed to paraffin embedding for hematoxylin and eosin (HE) staining by routine methods. The presence of H. pylori was assessed on gastric biopsies using Giemsa staining. Gastric pathology was recorded as per the Sydney system [34].
2.7DNA extraction and polymerase chain reaction (PCR)DNA extraction from gastric biopsies was performed according to the manufacturer's instructions (ADN PuriPrep-T kit, InbioHighway, Argentina). Samples were stored at −20 °C until used. PCR was performed by using specific primers. Target gene, amplicon size, primer names, and sequences are shown in Table 1. For PCR amplification, 50 ng of DNA samples was added to a PCR mixture containing 20 μmol forward and reverse primers, 15 μL of MINT Master Mix 2x (InbioHighway, Argentina) to the total volume of 25 μL. PCR amplification was performed under the following conditions: initial denaturation at 94 °C for 4 min followed by 35 cycles of denaturation at 95 °C for 30 s, annealing for 30 s (Table 1), extension at 72 °C for 30 s, and final extension at 72 °C for 5 min (Labnet MultiGene MiniThermocycler). The PCR reaction products were electrophoresed on 2 % agarose gel (InbioHighway, Argentina) and the bands were visualized by ethidium bromide staining. The cagA [35] and vacA [36] statuses were determined from H. pylori positive samples by PCR using their respective primers (Table 1).
Primer sets used for genotyping H. pylori virulence genes by PCR.
| Target site | Amplicon size (bp) | Primer names and sequences | Annealing temperature | References |
|---|---|---|---|---|
| cagA | 189 | cagA-F (5-TTGACCAACAACCACAAACCGAAG-3) | 62 °C | [35] |
| cagA-R (5-CTTCCCTTAATTGCGAGATTCC-3) | ||||
| vacAs1 | 259 | VA1-F (5-ATGGAAATACAACAAACACAC-3) | 56 °C | [36] |
| VA1-R (5-CTGCTTGAATGCGCCAAAC-3) | ||||
| vacAs2 | 286 | VA1-F (5-ATGGAAATACAACAAACACAC-3) | 56 °C | [36] |
| VA1-R (5-CTGCTTGAATGCGCCAAAC-3) | ||||
| vacAm1 | 290 | VA3-F (5-GGTCAAAATGCGGTCATGG-3) | 56 °C | [36] |
| VA3-R (5-CCATTGGTACCTGTAGAAAC-3) | ||||
| vacAm2 | 352 | VA4-F (5-GGAGCCCCAGGAAACATTG-3) | 56 °C | [36] |
| VA4-R (5-ATAACTAGCGCCTTGCAC-3) |
PCR primer sets, annealing temperature and size of the PCR products used for genotyping H. pylori. F – forward; R – reverse.
Sample size calculation was performed assuming a prevalence of non-alcoholic fatty liver disease in general population of 30 % [37] and 44.5 % in H. pylori infected subjects [38]. With 80 % of power and alpha level of 0.05, we calculated that at least 346 patients would be needed for the study.
Data were presented as mean ± SD, median (IQR, interquartile range), or number of subjects ( % of total) as appropriate. Differences between groups were analyzed by Students’ t-test or ANOVA for normal distribution, Wilcoxon rank sum test or Kruskal Wallis test for non-normal distribution. Categorical values were compared using Chi-square tests. The relationship with H. pylori and combined cagA/vacAs1/vacAm1 allelic carriers with VCTE cut-off to rule-out intermediate-high risk of SF/AF were examined by logistic regression model (binary response variable). Univariate models and multiple predictor variable models including age, gender, pre-DBT/type-II DBT, BMI and Hypertension as covariates were assessed. Data were analyzed using SPSS 22.0, and Jamovi 2.2.5. Two-tailed P < 0.05 was considered statistically significant.
2.9Ethical statementWritten informed consent was obtained from each participant enrolled in the study. The study protocol was approved by local ethical committee (Comité de Ética en Investigación Provincial, CEIP, N°454/17). The study was performed in accordance with the ethical standards as laid down in the 1975 Declaration of Helsinki and its later amendments.
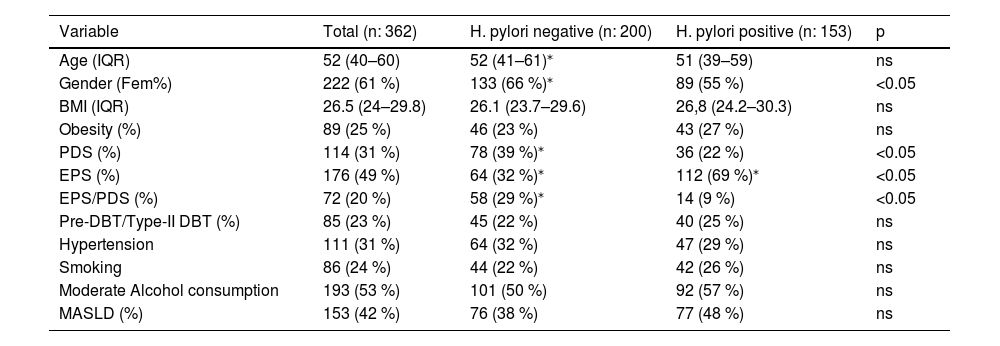
3Results3.1Study populationFour hundred and one patients with FD who met Rome-III criteria were evaluated before upper endoscopy, 39 patients were excluded and 362 patients were included for analysis. A flowchart of the study and baseline characteristics of patients appear in Fig. 1 and Table 2. The cohort of patients comprised of 222 (61 %) women and 140 (39 %) men with a median age of 52 (IQR, 40–60) years. The FD syndromes were as follows: 176 (49 %) patients had EPS, 114 (31 %) PDS and 72 (20 %) were EPS/PDS overlap. H. pylori positive gastric biopsies were detected in 162 (45 %). About clinical comorbidities: 85 (23 %) patients had pre-DBT/type-II DBT, 111 (31 %) hypertension, median BMI was 26.5 (IQR, 24–29.8) kg/m2 and 89 (25 %) had obesity (BMI ≥ 30 kg/m2), regular smoking was present in 86 (24 %) patients, 193 (53 %) subjects referred moderate alcohol consumption daily or occasional. The prevalence of MASLD was 153/362 (42 %) (Table 2). Viral serologic status of the study population was negative. Then, we evaluated our cohort's association of H. pylori infection and clinical variables. No differences were observed regarding H. pylori status at age, BMI, obesity, pre-DBT/type-II DBT, hypertension, smoking, and alcohol consumption (Table 2). H. pylori infection was significantly associated with male gender and EPS. While PDS and EPS/PDS was associated with H. pylori negative FD. A non-significant trend to more proportion of MASLD subjects was observed under H. pylori positive (77/162, 48 %, p 0.068) rather than H. pylori negative infection (76/200, 38 %).
, H. pylori negative, H. pylori positive, Non-alcoholic fatty liver disease (MASLD).")
H. pylori infection and clinical variables in FD subjects.
| Variable | Total (n: 362) | H. pylori negative (n: 200) | H. pylori positive (n: 153) | p |
|---|---|---|---|---|
| Age (IQR) | 52 (40–60) | 52 (41–61)⁎ | 51 (39–59) | ns |
| Gender (Fem%) | 222 (61 %) | 133 (66 %)⁎ | 89 (55 %) | <0.05 |
| BMI (IQR) | 26.5 (24–29.8) | 26.1 (23.7–29.6) | 26,8 (24.2–30.3) | ns |
| Obesity (%) | 89 (25 %) | 46 (23 %) | 43 (27 %) | ns |
| PDS (%) | 114 (31 %) | 78 (39 %)⁎ | 36 (22 %) | <0.05 |
| EPS (%) | 176 (49 %) | 64 (32 %)⁎ | 112 (69 %)⁎ | <0.05 |
| EPS/PDS (%) | 72 (20 %) | 58 (29 %)⁎ | 14 (9 %) | <0.05 |
| Pre-DBT/Type-II DBT (%) | 85 (23 %) | 45 (22 %) | 40 (25 %) | ns |
| Hypertension | 111 (31 %) | 64 (32 %) | 47 (29 %) | ns |
| Smoking | 86 (24 %) | 44 (22 %) | 42 (26 %) | ns |
| Moderate Alcohol consumption | 193 (53 %) | 101 (50 %) | 92 (57 %) | ns |
| MASLD (%) | 153 (42 %) | 76 (38 %) | 77 (48 %) | ns |
Data are expressed as median (IQR, interquartile range), or percentage (%) of the total. For continuous variables, Wilcoxon rank sum test was used. For categorical variables, Chi-square test was used.
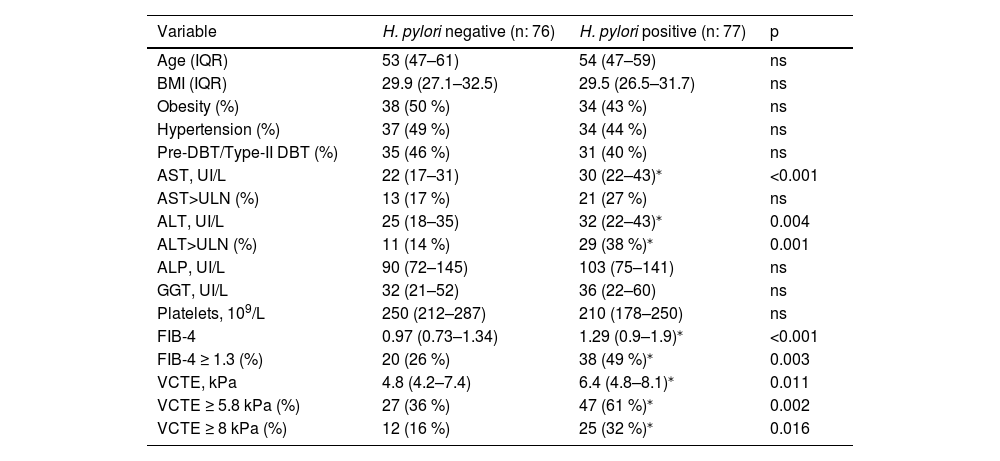
Then, we evaluate the influence of H. pylori status in non-invasive markers of liver injury and fibrosis in our cohort of FD patients with MASLD (n: 153) (Table 3). H. pylori active infection status showed no difference at age, BMI, obesity, pre-DBT/type-II DBT, hypertension, platelets, ALP and GGT values in MASLD subjects. Serum AST and ALT values resulted in a significant increase in H. pylori positive subjects (Table 3), though median values fall into de normal range. Next, to evaluate the clinical impact of AST and ALT in our cohort of MASLD, we used a cut-off of 40 IU/mL to categorize patients with upper limit of normal values (ULN). Indeed, more proportion of patients with ALT>ULN was attained in H. pylori positive but no difference at AST>ULN was observed (Table 3). To further evaluate liver injury, non-invasive markers of fibrosis — FIB-4 score and liver stiffness measurement by vibration control transient elastography (VCTE) — was determined. Consistent with AST and ALT levels data, FIB-4 and VCTE were significantly increased in H. pylori positive versus H. pylori negative subjects (Table 3). Furthermore, using cut-off values to rule-out of significant/advanced fibrosis (FIB-4 ≥ 1.3, VCTE ≥5.8 kPa and VCTE ≥8 kPa), we observed a significant trend of more proportion of H. pylori positive with FIB-4 ≥ 1.3 (38/77, 49 %, p 0.003), VCTE ≥5.8 kPA (47/77, 61 %, p 0.002) and VCTE ≥8 kPa (25/77, 32 %, p 0.016) compared to H. pylori negative patients (FIB-4 ≥ 1.3 20/76, 26 %; VCTE ≥5.8 kPa 27/76, 36 %, VCTE ≥8 kPa 12/76, 16 %) (Table 3). These data may indicate that MASLD subjects with H. pylori active infection have elevated markers of liver injury and fibrosis.
H. pylori infection in MASLD subjects.
| Variable | H. pylori negative (n: 76) | H. pylori positive (n: 77) | p |
|---|---|---|---|
| Age (IQR) | 53 (47–61) | 54 (47–59) | ns |
| BMI (IQR) | 29.9 (27.1–32.5) | 29.5 (26.5–31.7) | ns |
| Obesity (%) | 38 (50 %) | 34 (43 %) | ns |
| Hypertension (%) | 37 (49 %) | 34 (44 %) | ns |
| Pre-DBT/Type-II DBT (%) | 35 (46 %) | 31 (40 %) | ns |
| AST, UI/L | 22 (17–31) | 30 (22–43)⁎ | <0.001 |
| AST>ULN (%) | 13 (17 %) | 21 (27 %) | ns |
| ALT, UI/L | 25 (18–35) | 32 (22–43)⁎ | 0.004 |
| ALT>ULN (%) | 11 (14 %) | 29 (38 %)⁎ | 0.001 |
| ALP, UI/L | 90 (72–145) | 103 (75–141) | ns |
| GGT, UI/L | 32 (21–52) | 36 (22–60) | ns |
| Platelets, 109/L | 250 (212–287) | 210 (178–250) | ns |
| FIB-4 | 0.97 (0.73–1.34) | 1.29 (0.9–1.9)⁎ | <0.001 |
| FIB-4 ≥ 1.3 (%) | 20 (26 %) | 38 (49 %)⁎ | 0.003 |
| VCTE, kPa | 4.8 (4.2–7.4) | 6.4 (4.8–8.1)⁎ | 0.011 |
| VCTE ≥ 5.8 kPa (%) | 27 (36 %) | 47 (61 %)⁎ | 0.002 |
| VCTE ≥ 8 kPa (%) | 12 (16 %) | 25 (32 %)⁎ | 0.016 |
Data are expressed as median (IQR, interquartile range) or percentage (%) of the total. For continuous variables, the Wilcoxon rank sum test was used. For categorical variables, the Chi-square test was used. H. pylori, Helicobacter pylori; MASLD, metabolic associated steatotic liver disease; IQR, interquartile range; %, percentage; p, probability value.
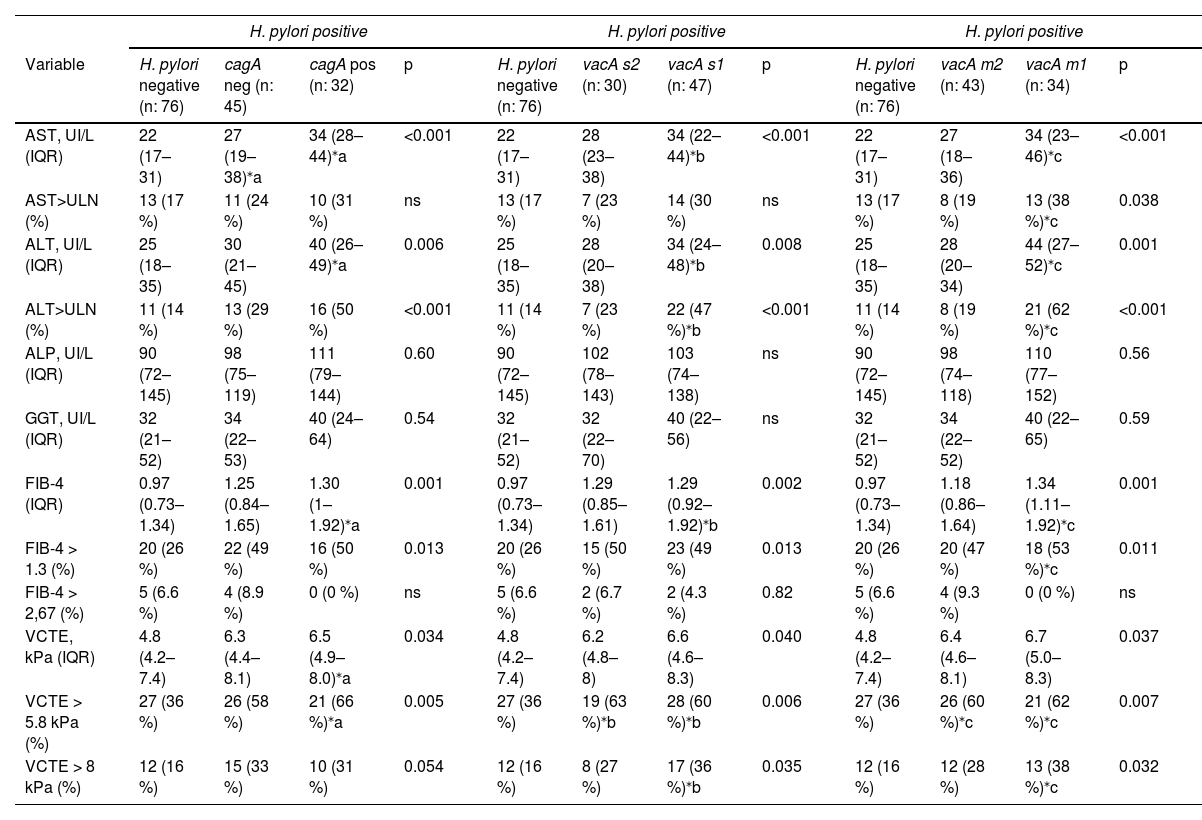
Next, we ascertained the influence of H. pylori virulence genes cagA and vacA from gastric biopsies on non-invasive markers of liver injury and fibrosis in our MASLD cohort. We first explored cagA, where 32 out of 77 (41 %) H. pylori positive gastric biopsies were carriers of cagA strain. Serum AST and ALT levels were significantly increased in cagA positive carriers, and no difference was observed at ALP nor GGT levels (Table 4). Likewise, using ULN cut-off analysis a significant more proportion of patients with ALT>ULN was attained in cagA positive carriers, but no difference was observed at AST>ULN (Table 4). Non-invasive markers of liver fibrosis, FIB-4 and VCTE, were increased in H. pylori cagA positive patients (Table 4). Furthermore, cagA carriers were associated with more proportion of patients with VCTE ≥5.8 kPa but not at VCTE ≥8 kPa (Table 4). More proportion of subjects with FIB-4 ≥ 1.3 was associated in H. pylori positive status irrespective of cagA carriage (Table 4).
H. pylori virulence genes cagA, vacA s1/s2 and m1/m2 in MASLD subjects.
| H. pylori positive | H. pylori positive | H. pylori positive | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variable | H. pylori negative (n: 76) | cagA neg (n: 45) | cagA pos (n: 32) | p | H. pylori negative (n: 76) | vacA s2 (n: 30) | vacA s1 (n: 47) | p | H. pylori negative (n: 76) | vacA m2 (n: 43) | vacA m1 (n: 34) | p |
| AST, UI/L (IQR) | 22 (17–31) | 27 (19–38)⁎a | 34 (28–44)⁎a | <0.001 | 22 (17–31) | 28 (23–38) | 34 (22–44)⁎b | <0.001 | 22 (17–31) | 27 (18–36) | 34 (23–46)⁎c | <0.001 |
| AST>ULN (%) | 13 (17 %) | 11 (24 %) | 10 (31 %) | ns | 13 (17 %) | 7 (23 %) | 14 (30 %) | ns | 13 (17 %) | 8 (19 %) | 13 (38 %)⁎c | 0.038 |
| ALT, UI/L (IQR) | 25 (18–35) | 30 (21–45) | 40 (26–49)⁎a | 0.006 | 25 (18–35) | 28 (20–38) | 34 (24–48)⁎b | 0.008 | 25 (18–35) | 28 (20–34) | 44 (27–52)⁎c | 0.001 |
| ALT>ULN (%) | 11 (14 %) | 13 (29 %) | 16 (50 %) | <0.001 | 11 (14 %) | 7 (23 %) | 22 (47 %)⁎b | <0.001 | 11 (14 %) | 8 (19 %) | 21 (62 %)⁎c | <0.001 |
| ALP, UI/L (IQR) | 90 (72–145) | 98 (75–119) | 111 (79–144) | 0.60 | 90 (72–145) | 102 (78–143) | 103 (74–138) | ns | 90 (72–145) | 98 (74–118) | 110 (77–152) | 0.56 |
| GGT, UI/L (IQR) | 32 (21–52) | 34 (22–53) | 40 (24–64) | 0.54 | 32 (21–52) | 32 (22–70) | 40 (22–56) | ns | 32 (21–52) | 34 (22–52) | 40 (22–65) | 0.59 |
| FIB-4 (IQR) | 0.97 (0.73–1.34) | 1.25 (0.84–1.65) | 1.30 (1–1.92)⁎a | 0.001 | 0.97 (0.73–1.34) | 1.29 (0.85–1.61) | 1.29 (0.92–1.92)⁎b | 0.002 | 0.97 (0.73–1.34) | 1.18 (0.86–1.64) | 1.34 (1.11–1.92)⁎c | 0.001 |
| FIB-4 > 1.3 (%) | 20 (26 %) | 22 (49 %) | 16 (50 %) | 0.013 | 20 (26 %) | 15 (50 %) | 23 (49 %) | 0.013 | 20 (26 %) | 20 (47 %) | 18 (53 %)⁎c | 0.011 |
| FIB-4 > 2,67 (%) | 5 (6.6 %) | 4 (8.9 %) | 0 (0 %) | ns | 5 (6.6 %) | 2 (6.7 %) | 2 (4.3 %) | 0.82 | 5 (6.6 %) | 4 (9.3 %) | 0 (0 %) | ns |
| VCTE, kPa (IQR) | 4.8 (4.2–7.4) | 6.3 (4.4–8.1) | 6.5 (4.9–8.0)⁎a | 0.034 | 4.8 (4.2–7.4) | 6.2 (4.8–8) | 6.6 (4.6–8.3) | 0.040 | 4.8 (4.2–7.4) | 6.4 (4.6–8.1) | 6.7 (5.0–8.3) | 0.037 |
| VCTE > 5.8 kPa (%) | 27 (36 %) | 26 (58 %) | 21 (66 %)⁎a | 0.005 | 27 (36 %) | 19 (63 %)⁎b | 28 (60 %)⁎b | 0.006 | 27 (36 %) | 26 (60 %)⁎c | 21 (62 %)⁎c | 0.007 |
| VCTE > 8 kPa (%) | 12 (16 %) | 15 (33 %) | 10 (31 %) | 0.054 | 12 (16 %) | 8 (27 %) | 17 (36 %)⁎b | 0.035 | 12 (16 %) | 12 (28 %) | 13 (38 %)⁎c | 0.032 |
Data are expressed as median (IQR, interquartile range), or percentage (%) of total. For continuous variables, Kruskal-Wallis test was used. For categorical variables, Chi-square test was used. For pairwise comparisons between groups Dwass-Steel-Critchlow-Fligner pairwise non-parametric test was used. (a), cagA positive or negative carriers versus H. pylori negative. (b), vacA-s1 or -s2 carriers versus H. pylori negative. (c), vacA-m1 or -m2 carriers versus H. pylori negative.
Then, we explored vacA-s1/-s2 and -m1/-m2, 47/77 (61 %) and 30/77 (39 %) of H. pylori positive gastric biopsies were carriers of vacA-s1 and -s2 strain, 34/77 (44 %) and 43/77 (56 %) were carriers of vacA-m1 and -m2 strain respectively. AST and ALT serum levels were significantly increased and more proportion of patients with ALT>ULN were associated with vacA-s1 carriers, no difference was observed at ALP, GGT levels and AST>ULN between groups (Table 4). In addition, vacA-m1 carriers showed a strong association with increased AST and ALT with more proportion of patients with ALT>ULN (62 %) and AST>ULN (38 %) (Table 4). Also, FIB-4 was increased in vacA-m1 carriers, but was similarly increased in vacA-s1 and -s2 groups. Higher liver stiffness by VCTE was associated with H. pylori infection regardless vacA-s1/-s2 and -m1/-m2 carrier status. Remarkably, carriers of vacA-s1 (36 %) and vacA-m1 (38 %) were associated with more proportion of patients with VCTE ≥8 kPa.
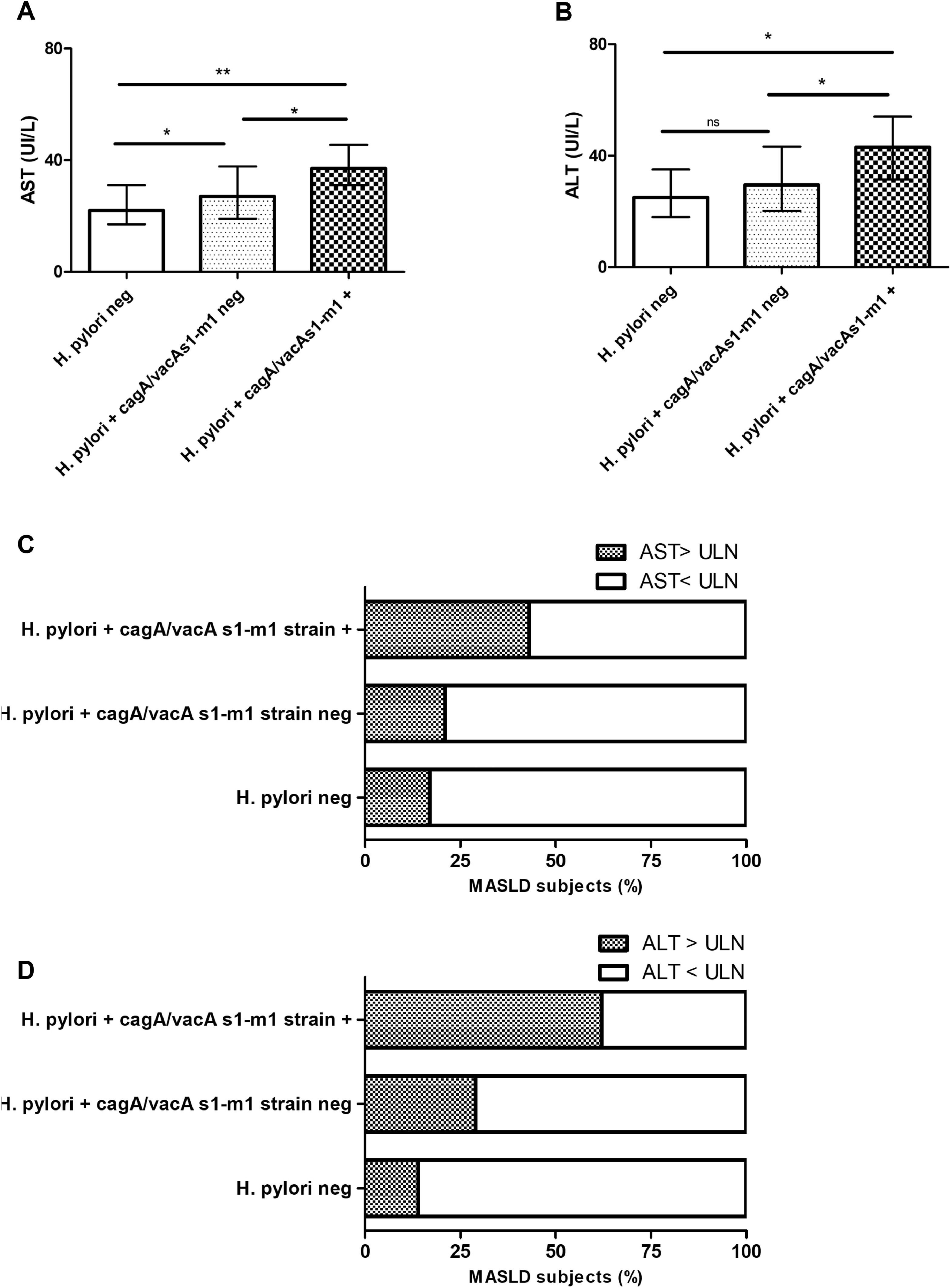
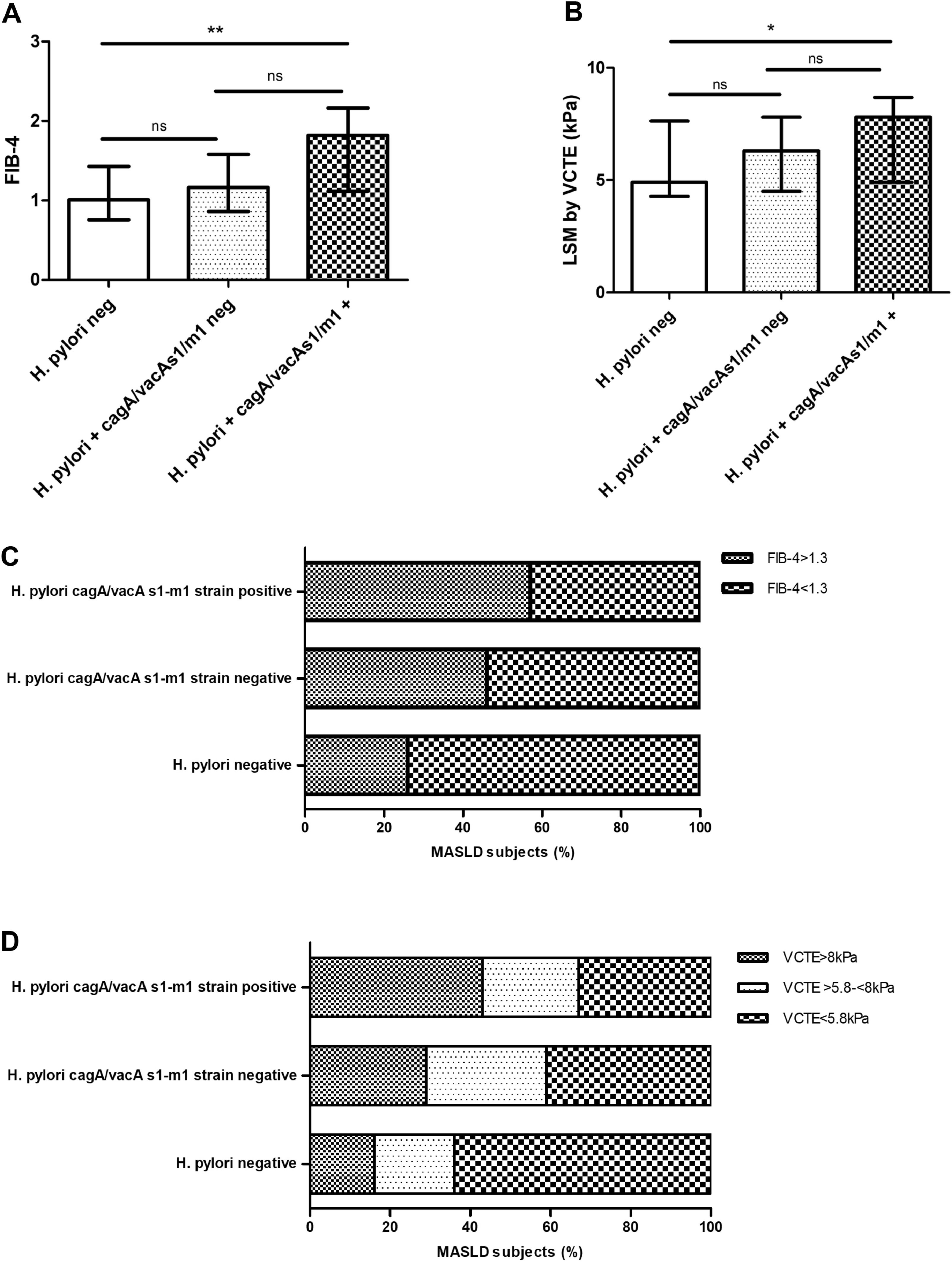
3.4H. pylori cagA/vacA-s1/m1-positive allelic carriers in MASLD subjectsNext, we evaluated the allelic combination analysis with cagA/vacA-s1/m1. A significant association with higher AST, ALT, AST>ULN (43 %, p 0.042) and ALT>ULN (62 %, p < 0.001) was attained in combined cagA/vacA-s1/m1-positive allelic carriers (Fig. 2A, B, C and D). Likewise, FIB-4 and LSM by VCTE was increased and a clear association with more proportion of patients with FIB-4 ≥ 1.3 (57 %, p 0.009), VCTE ≥ 5.8 kPa (67 %, p 0.006) and VCTE ≥ 8 kPa (43 %, p 0.023) was observed in combined cagA/vacA-s1/m1-positive allelic carriers (Fig. 3A, B, C and D).
 AST serum levels. B) ALT serum levels. C) AST > or < ULN. D) ALT > or < ULN. Data are expressed as median and interquartile range (IQR), and ( %) percentage of MASLD subjects. For continuous variables pairwise comparisons between groups Dwass-Steel-Critchlow-Fligner pairwise non-parametric test was used. For categorical variables, Chi-square test was used. * p < 0.05, ** p < 0.001.")
AST and ALT in patients with MASLD stratified according to H. pylori negative status, H. pylori positive cagA/vacA s1/m1-negative carrier and H. pylori cagA/vacA s1/m1-positive carrier. A) AST serum levels. B) ALT serum levels. C) AST > or < ULN. D) ALT > or < ULN. Data are expressed as median and interquartile range (IQR), and ( %) percentage of MASLD subjects. For continuous variables pairwise comparisons between groups Dwass-Steel-Critchlow-Fligner pairwise non-parametric test was used. For categorical variables, Chi-square test was used. * p < 0.05, ** p < 0.001.
![Non-invasive markers of high risk of significant/advanced fibrosis in patients with MASLD stratified according to H. pylori negative status, H. pylori positive cagA/vacA s1/m1-negative carrier and H. pylori cagA/vacA s1/m1-positive carrier. A) FIB-4 values. B) Liver stiffness measurement by vibration controlled transient elastography (LSM by VCTE). C) FIB-4 score ≥ 1.3 or < 1.3.. D) LSM by VCTE distribution by LSM cut-offs to rule-out significant fibrosis < 5.8 kPa, intermediate risk ≥5.8 kPa-<8 kPa, and to rule-out advanced fibrosis ≥8 kPa [31,32,33]. Data are expressed as median and interquartile range (IQR), and (%) percentage of MASLD subjects. For continuous variables pairwise comparisons between groups Dwass-Steel-Critchlow-Fligner pairwise non-parametric test was used. For categorical variables, Chi-square test was used. * p < 0.05, ** p < 0.001.](https://static.elsevier.es/multimedia/16652681/0000002900000006/v2_202411111025/S1665268124003351/v2_202411111025/en/main.assets/gr3.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNeKOJqKH5+Za6P+RA5Vq9DlYByYg1V9qqY5v8XuzTOGziiodjj66v0jlpEwEJN+eCTjoT02rGNxKttszavvnGnEtdH8Ffi/Rc8olxcqqwTbK3XttWbeEsuO+scGgBap/qjrLNUty9aRTXpRK/KaVC+64Er2AgrZ8aniI/4J6ntNlLDTlfbQbQcUsWMcNeu00Ak3EvdlGCQ/5qoXYxqnt2keH7+QorlTH2Y6yVqxOrM25w== "Non-invasive markers of high risk of significant/advanced fibrosis in patients with MASLD stratified according to H. pylori negative status, H. pylori positive cagA/vacA s1/m1-negative carrier and H. pylori cagA/vacA s1/m1-positive carrier. A) FIB-4 values. B) Liver stiffness measurement by vibration controlled transient elastography (LSM by VCTE). C) FIB-4 score ≥ 1.3 or < 1.3.. D) LSM by VCTE distribution by LSM cut-offs to rule-out significant fibrosis < 5.8 kPa, intermediate risk ≥5.8 kPa-<8 kPa, and to rule-out advanced fibrosis ≥8 kPa [31,32,33]. Data are expressed as median and interquartile range (IQR), and (%) percentage of MASLD subjects. For continuous variables pairwise comparisons between groups Dwass-Steel-Critchlow-Fligner pairwise non-parametric test was used. For categorical variables, Chi-square test was used. * p < 0.05, ** p < 0.001.")
Non-invasive markers of high risk of significant/advanced fibrosis in patients with MASLD stratified according to H. pylori negative status, H. pylori positive cagA/vacA s1/m1-negative carrier and H. pylori cagA/vacA s1/m1-positive carrier. A) FIB-4 values. B) Liver stiffness measurement by vibration controlled transient elastography (LSM by VCTE). C) FIB-4 score ≥ 1.3 or < 1.3.. D) LSM by VCTE distribution by LSM cut-offs to rule-out significant fibrosis < 5.8 kPa, intermediate risk ≥5.8 kPa-<8 kPa, and to rule-out advanced fibrosis ≥8 kPa [31,32,33]. Data are expressed as median and interquartile range (IQR), and (%) percentage of MASLD subjects. For continuous variables pairwise comparisons between groups Dwass-Steel-Critchlow-Fligner pairwise non-parametric test was used. For categorical variables, Chi-square test was used. * p < 0.05, ** p < 0.001.
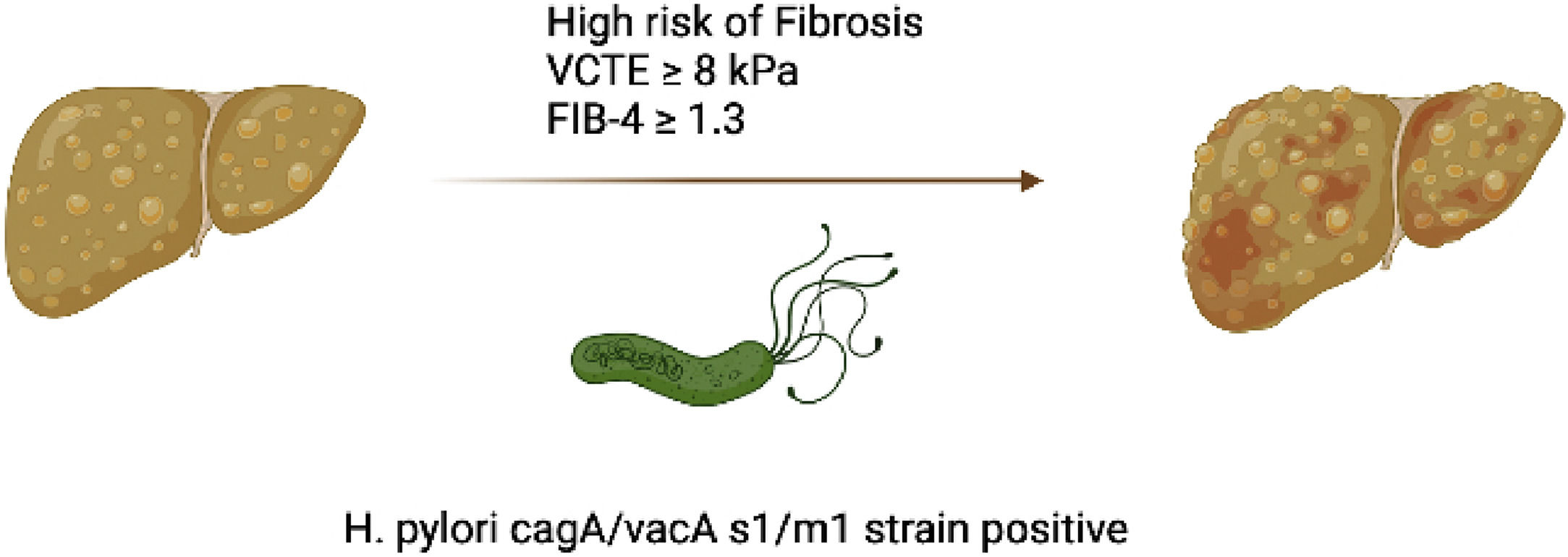
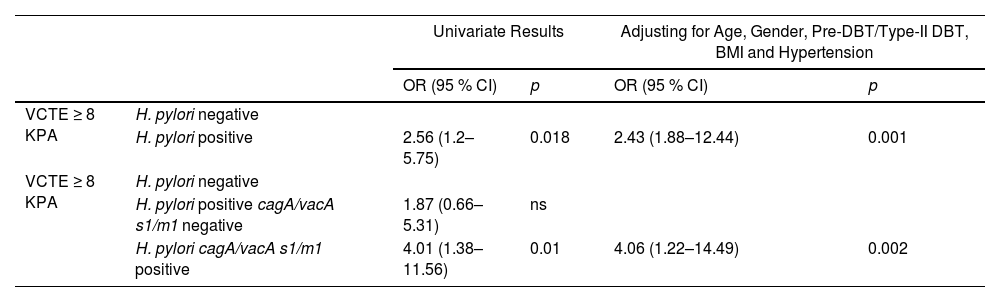
Since, fibrosis in MASLD is the most important prognostic factor and LSM by VCTE is one of the most validated non-invasive diagnostic tool for liver fibrosis evaluation [1,28]. We then, assessed by logistic regression model the impact of H. pylori status and combined cagA/vacAs1/vacAm1 allelic carriers on the VCTE ≥ 8 kPa cut-off to rule-out intermediate-high risk of SF/AF (Table 5). The OR for VCTE ≥ 8 kPa with H. pylori positive gastric biopsies was 2.56 (95 % CI, 1.2–5.75; p = 0.018) and for cagA/vacA-s1/-m1-positive allelic carriers was 4.01 (95 % CI, 1.38–11.56, p = 0.01), but non-significant association was observed by cagA/vacA-s1/m1-negative allelic carriers. After adjusting for age, gender, pre-DBT/type-II DBT, BMI and Hypertension the OR for VCTE ≥ 8 kPa with H. pylori positive gastric biopsies was 2.43 (95 % CI, 1.88–12.444; p = 0.009) and for cagA/vacA-s1/m1-positive allelic carriers was 4.06 (95 % CI, 1.22–14.49, p = 0.004). Collectively, these data suggest that, carriers of allelic combination cagA/vacA-s1/m1 are associated with markers of liver injury and may increase the risk of significant/advanced fibrosis in MASLD subjects (Fig. 4).
H. pylori, combined cagA/vacA s1/m1 carrier and VCTE ≥ 8 kPa.
OR: odds ratio, CI: confidence interval, ns: non-significant.
* p < 0.05.

The main findings of this observational cross-sectional study, pertain to the potential association of H. pylori virulence genes and non-invasive markers of severity in MASLD. Our results suggest that H. pylori infection in MASLD subjects is significantly associated with, (i) higher values of non-invasive markers of liver injury and fibrosis, (ii) carriers of cagA, vacA-s1and vacA-m1 showed increased values of AST, ALT, FIB-4 and LSM by VCTE, and (iii) carriers of H. pylori with cagA/vacA-s1/m1-positive allelic combination showed a strong association with elevated AST, ALT and independent risk of significant/advanced fibrosis by VCTE.
Ethiopathogenic association between H. pylori infection and MASLD is a matter of debate that was intended to be answered by various researcher worldwide from the bench, and clinical stand point [39,40]. First, is important to note that murine models of H. pylori infection do not have a fatty liver phenotype [41], but under a high fat diet H. pylori-positive mice had more hepatic steatosis and release of inflammatory mediators [41]. Noteworthy, in a cell cultures approach, H. pylori mediated hepatocyte injury is dependent upon cagA and vacA internalization [21,22]. Furthermore, in a liver fibrosis mice model using carbon tetrachloride, H. pylori-infected mice had increased fibrosis scores, and ALT and AST levels compared to uninfected mice [42]. For instance, local inflammation in the gastric and duodenal mucosa may lead to release of proinflammatory cytokines that increase gut permeability and access to portal circulation [17]. In this regard, H. pylori and other bacteria and toxins as well as cytokines may directly affect the hepatic parenchyma leading to and inflammatory milieu that may activate stellate cells to generate fibrosis [17]. A recent study, pointed toward an association between active H. pylori gastric infection and MASLD severity in morbidly obese patients subjected to bariatric surgery, in this study histological diagnostic “gold standard” for both main variables of interest (H. pylori gastric infection and MASLD) was used. Particularly, the rates of MASH by NAS score, as well as hepatic steatosis, inflammation, and fibrosis, were higher in H. pylori-positive than H. pylori-negative patients, however virulence genes status was not evaluated. Also, in study form China observed that H. pylori infection was not associated with steatosis but may be a risk factor for increased liver stiffness [38]. Our study showed a correlation with H. pylori-infection and elevated non-invasive markers of liver injury, and fibrosis by FIB-4 score and VCTE, with a strong association in carriers of cagA/vacA-s1/m1-positive allelic combination. Fibrosis is the most relevant prognostic factor in patients with MASLD [43]. We found a prevalence of high liver stiffness suggestive of significant/advanced fibrosis in 43 % of MASLD patients with H. pylori cagA/vacA-s1/m1-positive carriers. Our observation was further independent of indicators of disease severity or progression such as age, pre-DBT/type-II DBT, BMI and Hypertension [44]. Perhaps, the interaction between H. pylori and host genetic polymorphism delineates low-grade gastric and duodenal inflammation with eventually leading to hepatic pro-inflammatory milieu, and our results revealed that cagA/vacA-s1/m1-positive allelic combination strain significantly impact this observation. Since, the pathophysiology of MASLD is only partially revealed, it is considered as a multifactorial disorder, attributed to multiple or parallel “hits”, both genetic and environmental [6]. The precise mechanisms underlying the connection between H. pylori gastric infection and MASLD remain unclear. While the correlation between increased non-invasive markers of liver injury and fibrosis, H. pylori gastric infection and cagA/vacA-s1/m1-positive allelic combination strain does not prove causality. Confirmation from mechanistic studies is needed to clarify the clinical role of H. pylori virulence genes and fibrogenesis in MASLD. In this regard, the interaction between host genetic polymorphism background, H. pylori virulence genes and low-grade duodenal inflammation could explain high-risk of fibrosis in MASLD, and will be evaluated in our future research.
Second, multiple observational, prospective and metanalysis studies led to opposed conclusion on whether H. pylori infection contributes to MASLD [12,13] to no association between H. pylori infection and diagnosis of MASLD [14,15]. Our study failed to show an association between H. pylori infection and MASLD diagnosis by ultrasound. Perhaps, the prevalence of MASLD and H. pylori infection may not be the same in different regions, races, social customs, genetical background and eating habits, which makes the results of their correlation inconsistent. Therefore, the comprehensive analysis of data from multiple regions is needed. It is beyond the scope of this study to evaluate the impact of H. pylori eradication on MASLD non-invasive markers of liver injury and fibrosis. However, information is available from four clinical controlled studies [45,46,47,48]. While this four studies had disparate design, follow-up and end points making it difficult to compare, it seems that eradication therapy as adjunct to lifestyle intervention may improve non-invasive markers of steatosis, fibrosis and inflammation. Therefore, whether H. pylori eradication influences the occurrence or progression of MASLD is still controversial and needs to be confirmed by multicentric studies in the future.
The strengths of this study are that it is a prospective evaluation from a well-defined MASLD cohort. Endoscopists and Pathologists were blinded to medical history before evaluation. Non-invasive marker of liver fibrosis, LSM by VCTE was performed in all subjects. H. pylori virulence genes were genotyped in all H. pylori positive gastric biopsies. We choose FD patients for the current study because gastric biopsy sample is mostly available, which is the gold standard for H. pylori infection and virulence genes evaluation [49]. Also, FD etiopathogenesis is not related to MASLD and may not increase selection bias of the cohort [7,10].
The limitations of the study include: (1) population studied: Our cohort comprised of South-American population of functional dyspepsia, though we tried to investigate a cohort whose characteristics could resembled those of the general population, the modality of cohort recruitment did not allow us to affirm that our cohort was fully representative of the general population. However, our cohort was recruited independently from the hypothesis concerning high risk of MASLD severity. (2) Based on the non-invasive nature of the study design in FD-cohort, hepatic steatosis was detected by ultrasonography and fibrosis by VCTE but not liver histology, so the absence of histologic data prevents us from reporting the exact prevalence of steatohepatitis and advanced fibrosis according to the gold standard technique. Noteworthy, Noteworthy, VCTE is the most validated non-invasive method to accurately screen high risk of advanced fibrosis in MASLD [28,50]. Furthermore, LSM by VCTE can predict the occurrence of liver-related events [50].
5ConclusionsIn conclusion, the results of the current study suggest that in MASLD subjects with active H. Pylori infection is associated with elevated markers of liver injury and fibrosis, and carriers of cagA/vacA-s1/m1-positive allelic combination showed a higher risk of significant/advanced fibrosis by VCTE.
Author contributionsFJB contributed to the study concept and design, performed the statistical analysis, and drafted the manuscript, serving as the guarantor of the article. FJB and AS obtained funding. FM, MN, AS, and GV were involved in the acquisition of data as well as the analysis and interpretation of the data.
MVC, KE: acquisition of histological data, analysis and interpretation of data.
PDZ: analysis and interpretation of data, critical revision of the manuscript.
FundingFernando Javier Barreyro and Pedro Dario Zapata are supported by Scientific and Technical Research Council (CONICET) through a research mandate. The study is supported by a grant of the clinical research fund of Fundación HA Barceló.



