



Background & Aims. Juvenile hemochromatosis (JH) is a rare autosomal recessive disorder characterized by severe early-onset iron overload, caused by mutations in hemojuvelin (HJV), hepcidin (HAMP), or a combination of genes regulating iron metabolism. Here we describe two JH cases associated with simple heterozygosity for novel HJV mutations and unknown genetic factors. Case 1: A 12 year-old male from Central Italy with beta-thalassemia trait, increased aminotransferases, ferritin 9035 ng/ml and transferrin saturation 84%, massive hepatocellular siderosis and hepatic bridging fibrosis. Case 2: A 12 year-old female from Northern Italy with ferritin 467 ng/ml, transferrin saturation 87-95%, and moderate hepatic iron overload.
Material and methods. Direct sequencing of hemochromatosis genes (HFE-TfR2-HJV-HAMP-FPN-1) was performed in the children and siblings.
Results. In case 1, we detected heterozygosity for a novel HJV mutation (g.3659_3660insG), which was inherited together with the beta thalassemia trait from the father, who (as well as the mother) had normal iron parameters. In case 2, we detected another novel HJV mutation (g.2297delC) in heterozygosity, which was inherited from the mother, affected by mild iron deficiency. The father had normal iron stores. Both mutations are frameshifts determining premature stop codons. No other disease causing variant was detected.
Conclusion. Although beta-thalassemia trait was a possible cofactor of iron overload in case 1, iron overload cannot be explained by simple heterozygosity for HJV mutations in both cases. Other genetic factors should be investigated, and further studies are needed to understand genotype-phenotype correlations in JH.
Juvenile hemochromatosis (JH) is a rare autosomal recessive disorder characterized by severe iron overload presenting in the 2nd/3rd decade of life.1–6 Common clinical features include hypogonadotropic hypogonadism and cardiomyopathy, which are more common than in adult onset hereditary hemochromatosis (HH), which most frequently presents with hepatic iron overload or arthropathy and his related to homozygosity for C282Y Hemochromatosis gene(HFE) variant, which Mutations causing JH have been identified in the Hemojuvelin (HJV)1 and in the Hepcidin (HAMP) genes,2 or in a combination of these with other adult-onset HH genes (HFE, transferrin receptor-2: TfR2, Ferroportin-1: FPN1).3 Here we report two atypical JH cases, only partially explained by novel loss-of-function HJV mutations.
Material and MethodsDirect sequencing of coding sequences of HH causing genes (HFE, TfR2, HJV, HAMP and FPN1) and HAMP promoter was performed in the probands and their siblings. Written informed consent was obtained from each subject or guardian included in the study. The study was conducted according to the principles contained in the Declaration of Helsinki and approved by the Ethical Commitees of the Fondazione Ca’ Granda IRCCS Milano and of the Ospedale Bambin Gesù Roma.
Case oneA 12 year-old male was referred for suspected HH because of the incidental finding of high levels of aminotransferases and iron parameters. Parents were non-consanguineous and respectively coming from the Centre and South of Italy.
Blood tests showed microcytic anemia (Hb 10.2 g/ dL, MCV 70 fl), increased HbA2 concentrations (5.3%), consistent with beta-thalassemia trait, serum iron 238 mg/dL, transferrin 200 mg/dL, ferritin 9035 ng/mL, transferrin saturation 84%, reticulocytes 1.2%, alanine and aspartate aminotransferases 86/114 IU/mL. He resulted negative for hepatotropic viruses, celiac disease and infections; ceruloplasmin and alpha-1 antitrypsin levels were in the normal range, whereas anti-nuclear antibodies were present at low titres. Echocardiography was unremarkable, while abdominal ultrasonography demonstrated hepatosplenomegaly. The patient did not show any clinical, imaging, or laboratory evidence of arthrop- athy, hypogonadotropic hypogonadism or diabetes mellitus.
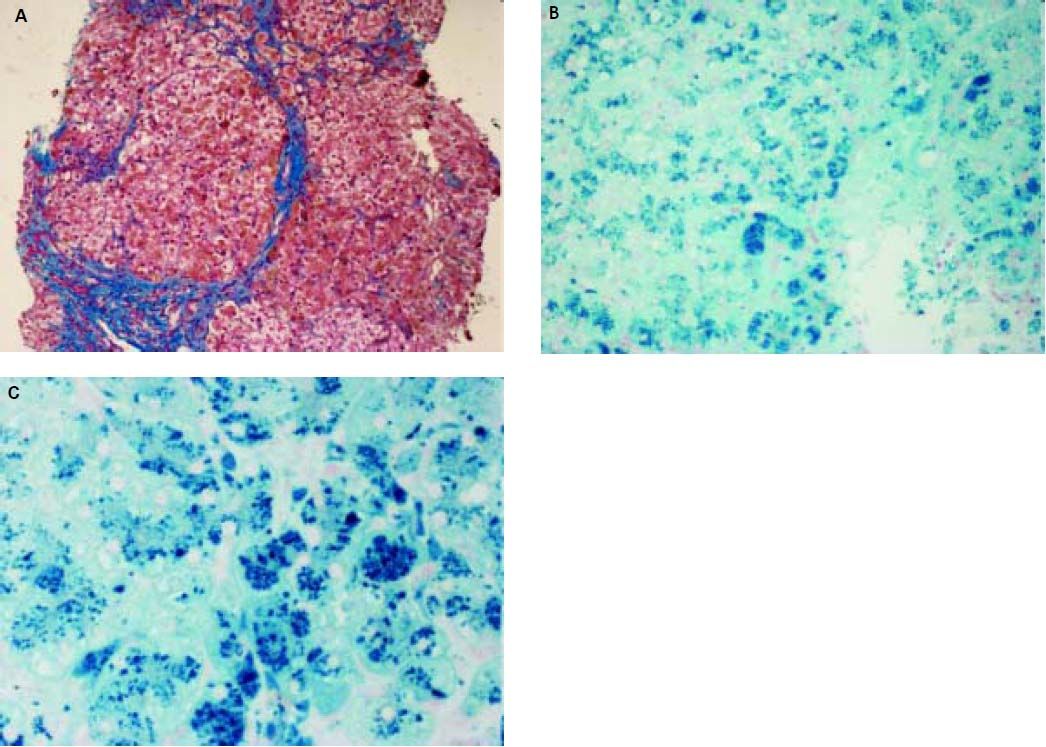
Histopathological evaluation of the liver demostrated uncomplete nodular architecture (Figure 1A) and massive parenchymal siderosis (Scheuer grade IV) with porto-central lobular gradient (HH pattern, pre-cirrhotic state, suggestive of JH; Figure 1B).
. B. massive parenchymal siderosis (Scheuer grade IV) with lobular gradient (hereditary hemochromatosis pattern, pre-cirrhotic state (Perls’ stain, iron deposition in haemosiderin is shown in blue; original magnification 10x). C. Perls’ staining at higher magnification (40x), supporting predominantly hepatocellular iron overload.")
Liver histology of case 1. A. Uncomplete nodular architecture with almost complete fibrous septa (hematoxylin & eosin, original magnification 10x). B. massive parenchymal siderosis (Scheuer grade IV) with lobular gradient (hereditary hemochromatosis pattern, pre-cirrhotic state (Perls’ stain, iron deposition in haemosiderin is shown in blue; original magnification 10x). C. Perls’ staining at higher magnification (40x), supporting predominantly hepatocellular iron overload.
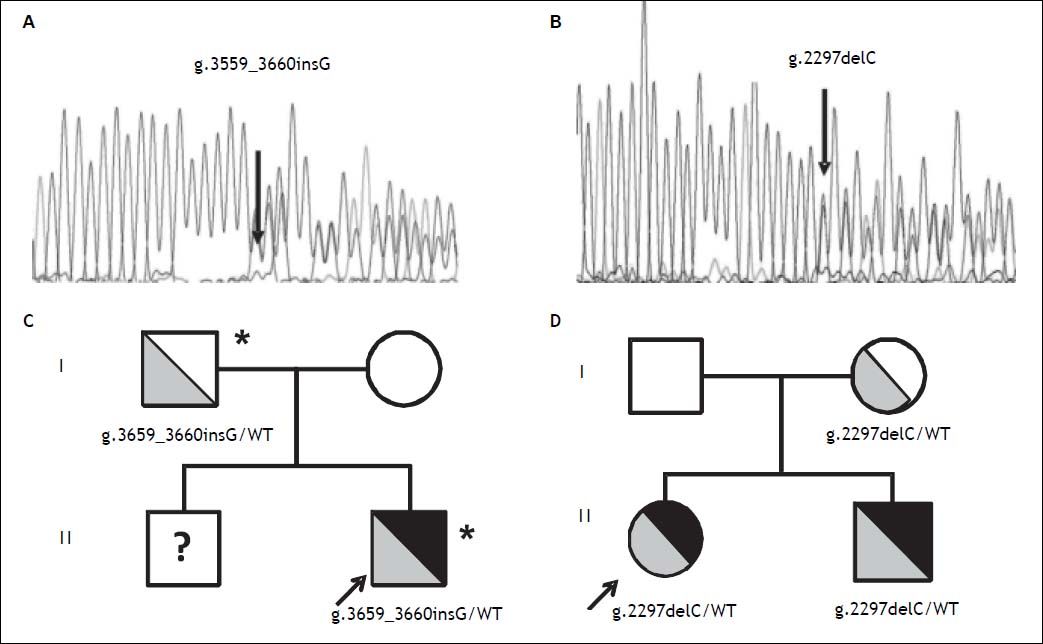
Direct sequencing of HH causing genes was performed and the g.3659_3660insG mutation in the HJV gene (encoding for a guanine insertion at position g.3659_3660, determining a frameshift with a premature stop codon p.Cys321Valfs) was detected in heterozygosity (Figure 2A). The mutation was inherited from the father, as for the beta thalassemia trait (Figure 2C). Both parents had normal iron parameters. The proband had an older brother who could not be evaluated, as he was under treatment for acute lymphoblastic leukemia. No other disease causing variant was detected. He was started on phlebotomy (250 mL every two weeks) until iron depletion. Iron depletion was however difficult to obtain because of the presence of the beta-thalassemia trait. After removal of about 3.5 g of iron, ferritin levels are still elevated (3603 ng/mL, transferrin saturation still 90%), even if aminotransferases went down upper normal ranges. He is continuing iron depletion.
, iron overload phenotype is shown in black; * beta-thalassemia trait. WT: Wilde type; ? unknown genotype.")
Transmission of novel HJV mutations. Electropherograms of novel HJV mutations. A. G insertion at position 3659-3660 detected in heterozygosity in case 1; p.Cys321Valfs. B. C deletion at position 2297 detected in heterozygosity in case 2; p.Gly102Glyfs. Family trees. C. Pedigree of case 1. D. Pedigree of case 2. HJV mutations are shown in grey (with genotypes listed below), iron overload phenotype is shown in black; * beta-thalassemia trait. WT: Wilde type; ? unknown genotype.
A 12 year-old female from Northern Italy was incidentally discovered with an isolated increase in iron parameters (ferritin 467 ng/mL, transferrin saturation 87%, normal blood count and aminotransferases). The patient showed normal statural growth for her age and was in good health; she had not reached menarche. Parents were non-consanguineous from Northern Italy; the maternal grandfather had died of hepatocellular carcinoma. Evaluation of hepatic iron by T2* MRI revealed moderate iron overload (285 mM) while cardiac MRI and cardiac iron concentration were normal. Genetic tests pointed out another novel nonsense mutation in the HJV gene (g.2297delC, determining a frameshift with a premature stop codon; p.Gly102Glyfs Figure 2B), present in heterozygosity. The mutation was inherited from the mother, who had mild iron deficiency, and was also present in the younger eight-year-old brother showing increased iron parameters (transferrin saturation 90%) and mild hepatic iron overload at MRI (Figure 2D). The patient was negative for mutations in other HH causing genes. She was started on phlebotomy (100 mL twice monthly, because of the very low weight: < 30 kg ), which were well tolerated. After removal of about 2 g or iron, ferritin levels are still elevated at 215 ng/mL and transferrin saturation is 95%. She is continuing iron depletion, but at age 13½ years has still not reached the menarche.
DiscussionHJV gene encodes for a protein of 426 aminoacids, which includes a C-terminal glycosylphosphatidyli- nositol anchor in hepatocytes. The membrane form of HJV represents the main driver of the expression of hepcidin, the hormone that inhibits iron absorption and recycling by binding the iron exporter FPN-1. Defective forms of HJV result in decreased hepcidin transcription and consequent iron overload.4 Various mutations in the HJV gene have been described. In particular, the HJV p.G320V mutation has been detected in 50% of families with JH.5,6
The g.3659_3660insG and the g.2297delC (pCys321Valfs and p.Gly102Glyfs) are two novel HJV mutations described in this report. The p.Cys321Valfs variant (corresponding to g.3659_3660insG genomic mutation) causes a frameshift leading to a truncated protein of 340 amino acids, while the p.Gly102Glyfs one (corresponding to g.2297delC mutation) generates a truncated protein of 112 amino acids. In both cases the GPI anchor, located at the end of exon 4 (residue Asp400), is lost, indicating that mutant proteins cannot be expressed at membrane level to induce hepcidin.
Besides, it has previously been shown that other HJV mutations causing truncation of the protein more distally than the present ones6 are causally linked to JH, strongly suggesting that these novel variants represent complete loss-of-function mutations.
Interestingly, in case 1 there was no evidence of cardiomyopathy, skin pigmentation or hypogonadotropic hypogonadism, typical of JH, but organ involvement was limited to liver disease. Primary amenorrhea could not be excluded in case 2.
The beta-thalassemia trait is a possible cofactor influencing iron overload and liver disease in case 1,7 but iron overload could not be explained by simple heterozygosity for JH in both cases.
Considering that a single mutation in the HJV gene (with wild-type HFE, TfR2, HAMP and FPN1 genotypes) has not been previously associated to such a severe iron overload (consistent with the autosomal recessive inheritance pattern of HH), and that an identical genetic status was not associated with iron overload in the parents of the probands, these two cases indicate that other types of genetic variants (large deletions, mutations in non-coding DNA regions) and/or other genetic loci contribute to the pathogenesis of HH. All children and parents reported standard diet, children were not supplemented with iron, and alcohol abuse was carefully excluded despite the young age in each case. However, we cannot completely exclude the possibility that unknown acquired factors may have played a role in the pathogenesis of iron overload in the probands.
Large collaborative studies exploiting next generation sequencing in patients and functional experiments are needed to better understand genotype-phenotype correlations in JH.
Abbreviations- •
FPN1: ferroportin-1.
- •
HAMP: hepcidin.
- •
HFE: hemochromatosis gene.
- •
HH: hereditary hemochromatosis.
- •
HJV: hemojuvelin.
- •
JH: juvenile hemochromatosis.
- •
TfR2: transferrin receptor-2.
There is no conflict of interest to disclose.
Financial SupportThe work was supported by the “Centro universitario per lo studio delle malattie metaboliche del fegato” (Academic research center) and by the “Associazione per lo studio e la cura delle malattie metaboliche del fegato (AMMF) ONLUS” (Nonprofit organization for the study and cure of metabolic liver diseases).



