






The purpose of the present work was to study the pharmacokinetics of ketorolac, a poorly metabolized drug, in experimental cirrhosis. Cirrhosis was induced by bile duct ligation (BDL) for four weeks in male Wistar rats. Ketorolac was given intravenously (1 mg/ kg) or orally (3.2 mg/kg) to control (sham-operated) and BDL-rats. Determination of ketorolac in plasma was carried out by HPLC and estimation of pharmacokinetic parameters was performed by non-compartmental analysis. Indicators of liver damage and liver fibrosis were significantly increased (p < 0.05) in BDL compared to control rats. Experimental cirrhosis did not induce any significant alteration in intravenous ketorolac pharmacokinetics. Volume of distribution, clearance, AUC and t1/2 were similar in BDL and control animals. Notwithstanding, oral ketorolac bioavailability was significantly altered in BDL rats. AUC and Cmax were reduced, while tmax was prolonged, suggesting that both, the extent and the rate of ketorolac absorption were decreased. Results show that liver cirrhosis may result in significant pharmacokinetic alterations, even for poorly bio-transformed drugs, but that alterations may vary with the route of administration. In conclusion, uncritical generalizations on the effect of liver damage on drug kinetics should be avoided and systematic studies for every drug and every route of administration are thus recommended.
Abbreviations
BDL bile duct ligation
HPLC high performance liquid chromatography
AUC area under the curve
t1/2 terminal half-life
Cmax peak concentration
tmax time to reach Cmax
NSAID non-steroidal anti-inflammatory drug
Vd volume of distribution
Cl total drug clearance
F absolute bio-availability after the oral route
γ-GTP γ-glutamyltranspeptidase
ALT alanine aminotranspeptidase
Grants
This study was supported in part by grant 38940-M from Conacyt, Mexico. Liliana Rivera-Espinosa and Mónica Ordaz-Gallo were fellows of Conacyt.
IntroductionKetorolac is a potent non-steroidal anti-inflammatory drug (NSAID) widely used for the treatment of moderate to severe pain, and it has been shown to be effective after administration by the oral, intramuscular and intravenous routes.1,2 As do other NSAIDs, ketorolac produces its effect through the inhibition of prostaglandin synthesis.3 However, due to its high potency and efficacy, additional mechanisms of action have been proposed, including a participation of opioid receptors,4 the stimulation of the L-arginine-nitric oxide-cyclic GMP pathway,5 and potassium channel opening.6 Using an experimental model with a noxious stimulus of constant intensity,7 reported that antinociceptive effect of ketorolac is directly related to its blood concentration. Notwithstanding, clinical studies in postoperative pain have shown that there is no direct relation between ketorolac plasma concentrations and its analgesic effect.8,9 The lack of direct correlation between plasma concentration and the effect has been attributed to the rapid variation in pain intensity occurring immediately after surgery. Plasma levels can be related to the analgesic response, however, by considering a placebo effect including a reduction in pain intensity during the first hours of the postoperative period.8 It then appears that, either directly or indirectly, the analgesic response to ketorolac depends on its pharmacokinetics. Therefore, any change in ketorolac absorption, distribution and/or elimination will likely have an impact on its pharmacological response.
There are some concerns on ketorolac side effects. Ketorolac, as other NSAIDs, may produce gastrointestinal side effects including peptic ulcer and bleeding.2,10 However, the main concern relates to its effect on kidney function, particularly in hypovolemic patients.11 There are several reports on acute renal failure observed with ketorolac, even after a single dose administration.12,13 Life-threatening reactions have been reported with doses of 60 mg or higher. Thus, the use of ketorolac doses higher than 30 mg is not recommended.1,9 Ketorolac then appears to be a drug with a relatively narrow therapeutic index. Hence, alterations in its bioavailability may result in a lack of efficacy or in toxicity. Situations such as hepatic and renal impairment may alter drug bioavailability.14 Despite of the wide use of ketorolac, the information available in the scientific literature on its pharmacokinetics in liver damage is scarce, and it is limited to a single published abstract on which patients with alcoholic cirrhosis are compared with young healthy individuals.15 According to this report in patients with cirrhosis, ketorolac half-life was slight, but significantly prolonged after intra-muscular administration, but there was no significant change in clearance. After oral administration, the time to reach the maximum concentration was prolonged in patients with cirrhosis, although no other oral bio-availability parameter was altered. Hence, it appears that the actual role of liver damage on ketorolac pharmacokinetics is not yet clear. Ketorolac is mainly eliminated by renal excretion of the unchanged drug.2,16 Thus, it can be assumed that liver damage will produce little change on its disposition due to the small role played by bio-transformation. Liver damage, however, may alter pharmacokinetics if only by impairing drug metabolism. Drug distribution can be altered by changes in drug binding to plasma proteins due to a reduced albumin production.17 Liver damage also may result in alterations in blood flow in the portal vein,18-20 which may modify the extent and rate of drug absorption from the gastrointestinal tract.
Systematic studies on the role of hepatic diseases on drug kinetics are difficult to perform in humans due to the extremely high interindividual variability found in the clinical situation. Besides, experimental animal models have shown to be a suitable alternative for the characterization of the pharmacokinetic alterations induced by liver damage.17,21 Therefore, the purpose of this work was to evaluate the effect of experimental liver cirrhosis on the pharmacokinetics of ketorolac after both, intravenous and oral administration.
Materials and methodsMaterials. Ketorolac tromethamine was obtained from Laboratorios Liomont S.A. (Mexico City). Sodium tolmetin was obtained as a sample from Cilag de México, S.A. (Mexico City). Acetonitrile, chromatographic grade, was purchased from Merck (Darmstadt, Germany). All other reagents used were of analytical grade.
Animals. Male Wistar rats (200-250 g) from our own breeding facilities were used in this study. All animals received human care and the study complied with the institution’s guidelines and the Mexican official regulation regarding technical specifications for production, care and use of laboratory animals (NOM-062-ZOO-1999).22 Additionally, the protocol followed the guidelines of the Canadian Council on Animal Care.23
Experimental cirrhosis. The cirrhosis was produced by prolonged bile duct ligation (BDL) in the rat, which is a widely used model to simulate the condition of cirrhosis in humans.24,25 Extrahepatic cholestasis was induced by the double ligation and section of the common bile duct, as described previously.24 Control sham-operated animals were submitted to all surgical procedures, but without obstruction of the bile duct. Twenty-eight days after surgery, liver damage was assessed by the plasma activities of γ-glutamyltranspeptidase (γ-GTP)26 and alanine aminotransferase (ALT).27 Small liver sections fixed in formalin were used for Mallory trichromic staining for histological examination under light microscopy. Collagen content, a marker of fibrosis, was estimated by the determination of hydroxyproline in fresh liver samples, as described previously.28
Study design. On the 28th day after BDL, food was withheld but animals had free access to drinking water. Under light ether anesthesia, polyethylene catheters (a combination of a PE-10 and PE-50, i.d. 0.28 mm, o.d. 61 mm and i.d. 0.58 mm, o.d. 0.96 mm, respectively, Clay Adams, Parsippany, NJ, USA) were implanted into the caudal artery to collect blood samples. For animals which received the drug by the intravenous route, a polyethylene catheter (PE-50) was implanted in a femoral vein and 1 mg/kg ketorolac was administered as a bolus dissolved in isotonic saline. For animals receiving the oral drug, 3.2. mg/kg ketorolac dissolved in saline was given by gavage. Ketorolac was administered after a 12 h fasting period. The used doses were selected as it has been shown that both, the intravenous and the oral doses yield comparable analgesic effects in experimental pain.7
Blood samples (0.2 mL) were drawn at 0, 10, 15, 30, 60, 120, 240, 360 and 480 min (drinking water was given during this process) after ketorolac administration and plasma was obtained by centrifugation. Plasma samples were stored at -70 °C until analysis. Once blood sampling was finished, animals were anesthetized with ether and submitted to exsanguination by cardiac puncture. This blood was used to determine enzyme markers of liver damage. Finally, animals were sacrificed by an excess of anesthesia and the liver was immediately removed to estimate hepatic collagen content.
Determination of ketorolac in plasma. Ketorolac concentration in plasma samples was determined using a high-performance liquid chromatographic method with UV detection, as described by Flores-Murrieta et al.29 Briefly, 0.4 mL of 0.05 M potassium phosphate buffer (pH= 7.4) and 100 ng of sodium tolmetin (internal standard) were added to 0.1 mL plasma samples. Samples were then acidified by the addition of 0.1 mL of 0.5 M solution of sodium acetate (pH=4) and extracted with 1 mL diethyl ether by vortex agitation during 1 min at maximum speed. The two phases were separated by centrifugation at 10,000 rpm for 5 min. The organic layer was transferred to a clean conical glass tube and evaporated to dryness at 50 °C under a gentle nitrogen stream. The dry residue was redissolved in 0.1 mL of deionized water and 40 μL aliquots were injected into the chromatographic system. Analyses were performed using a Novapak C-18 column (150 x 3.9 mm I.D., particle size 4 μm, Waters Assoc., Milford, MA, USA) eluted with a mixture of acetonitrile and phosphoric acid 1 mM (pH=3) 43:57 v/v at a constant flow rate of 1.4 mL/min and at room temperature. The effluent from the column was monitored at 313 nm. Retention times were 2.8 and 4.0 min for ketorolac and tolmetin, respectively.
Analysis of results. Individual plasma concentration versus time plots were constructed. For the intravenous route, the area under the curve (AUC), the volume of distribution (Vd), the total drug clearance (CL) and the terminal half-life (t1/2) were determined. AUC was estimated by the trapezoidal rule and extrapolated to infinity by multiplying the last detectable concentration by the time constant of the terminal concentration decay phase. Vd was obtained by dividing the dose by the extrapolated concentration corresponding to time zero. Clearance was estimated by dividing the dose by AUC. Half-life was estimated from the slope obtained by linear regression of the terminal phase of semilogarithmic concentration versus time plots. For the oral route, the peak concentration (Cmax), the time to reach this peak (tmax), and AUC were estimated. Cmax and tmax were determined graphically from concentration versus time plots. AUC was estimated as above. All pharmacokinetic parameters were obtained by non-compartmental analysis using the Win NONLIN software program (Pharsight Corp., Mountain View, CA, USA). In estimation of absolute bioavailability after the oral route (F) was calculated as: F = (AUCORAL/AUCIV) (DOSEIV/DOSEORAL) using mean values, since the intravenous and oral experiments were performed in different animals.14 Vd and CL were not estimated for the oral route as F values for individual animals were not available.
Results are presented as mean ± SEM. Comparisons between groups were performed using the student t test for unpaired data. Differences were considered to achieve statistical significance when p < 0.05. Since intravenous and oral pharmacokinetics were determined in different animals, F was estimated from mean AUC values. Hence, no SEM values are provided and no statistical test was performed to compare the values of this parameter between groups.
ResultsAfter four weeks, bile duct obstruction shown a pronounced liver damage. Fibrosis was evaluated by a histological approach. Prolonged biliary obstruction was accompanied by a marked increase in collagen disposition around the portal triad. The normal architecture was lost, extended necrotic areas were frequently observed and a marked ductular proliferation was present (Figure 1, right panel) with regard to sham operated animals (Figure 1, left panel). Plasma γ-GTP augmented about 14-times (Figure 2A) whereas ALT exhibited only a two-fold increase (Figure 2B). Hepatic collagen content, estimated as the hydroxyproline accumulation, was about six times higher in DBL than in sham-operated rats (Figure 2C). All the differences in liver damage markers between BDL and sham-operated animals achieved statistical significance (p < 0.05). All of the bile-duct blocked animals exhibited ascites at the end of the fourth week.

, alanine aminotransferase (ALT; panel B) determined in plasma and liver collagen content express as hydroxyproline (panel C) from sham-operated rats (SHAM) and bile duct ligated rats (BDL) Each bar represents the mean value of experiments performed in duplicate assays with samples from 12 animals ± SEM. (*) Statistically significantly different from the SHAM group, P < 0.05.")
Enzyme activities of γ-glutamyl transpeptidase (γ-GTP; panel A), alanine aminotransferase (ALT; panel B) determined in plasma and liver collagen content express as hydroxyproline (panel C) from sham-operated rats (SHAM) and bile duct ligated rats (BDL) Each bar represents the mean value of experiments performed in duplicate assays with samples from 12 animals ± SEM. (*) Statistically significantly different from the SHAM group, P < 0.05.
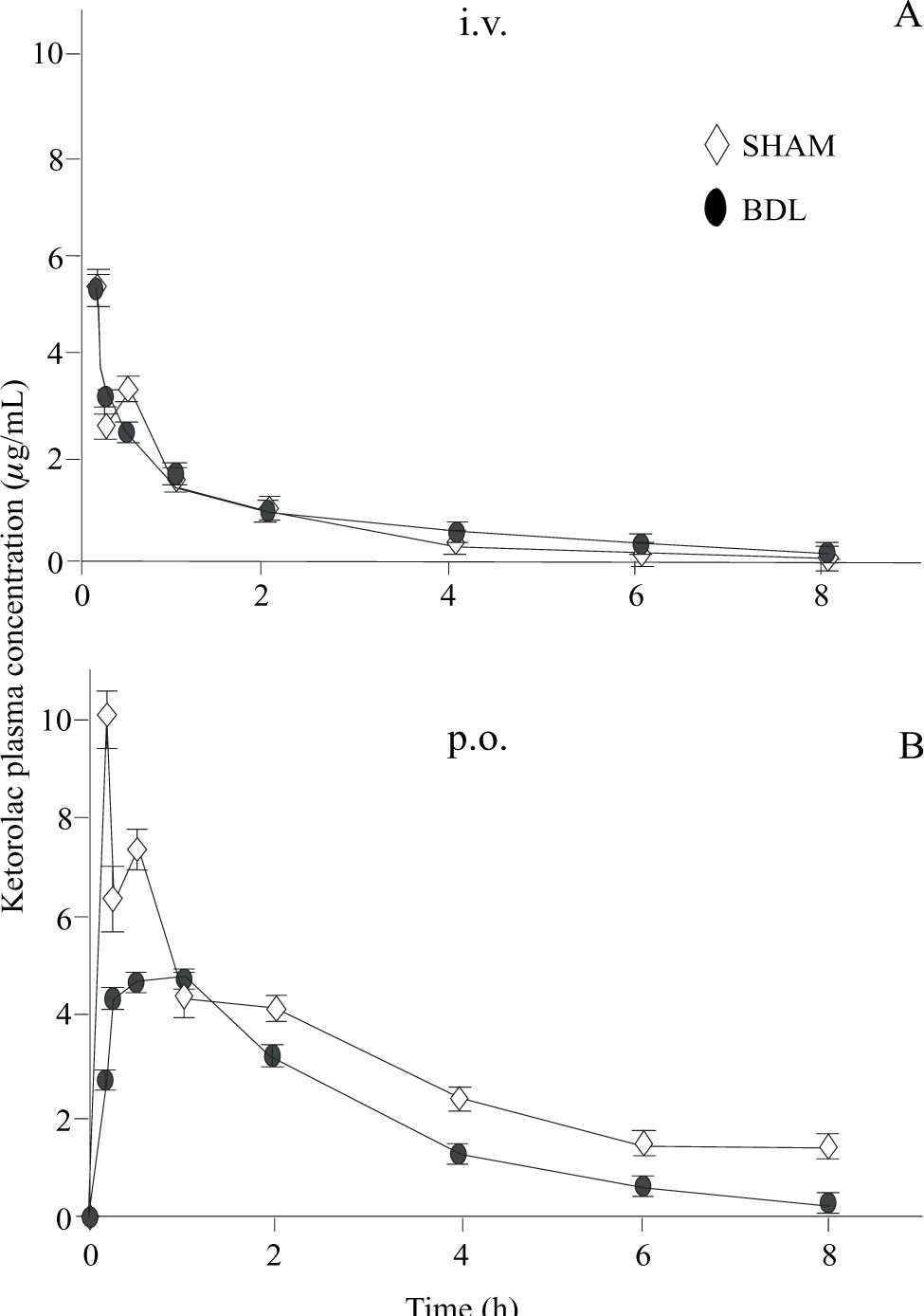
Plasma ketorolac concentrations following intravenous and oral administration are depicted in figure 3. Pharmacokinetic parameters obtained by non-compartmental analysis are shown in table I. BDL did not induce any significant change in ketorolac pharmacokinetics with regard to sham-operated animals when administered by the intravenous route. Notwithstanding, BDL was able to alter oral ketorolac bioavailability. AUC and Cmax were significantly reduced (p < 0.05) whereas tmax was significantly prolonged (p < 0.05). As a result of the AUC reduction after oral administration, without any change by the intravenous route, the absolute bioavailability (F), which was nearly complete in sham-operated animals, was reduced in BDL rats.
 dose (panel A) and after administration of a single oral (p.o.) 3.2. mg/kg dose (panel B) to shamoperated (◊) and bile duct ligated (DBL) rats (•) Data are presented as the mean ± S.E.M of 6 animals.")
Ketorolac plasma concentrations observed after administration of a single 1 mg/kg intravenous (i.v.) dose (panel A) and after administration of a single oral (p.o.) 3.2. mg/kg dose (panel B) to shamoperated (◊) and bile duct ligated (DBL) rats (•) Data are presented as the mean ± S.E.M of 6 animals.
Pharmacokinetic parameters of ketorolac observed after administration of a single 1 mg/kg intravenous dose and a single oral 3.2 mg/kg dose to rats with experimental cirrhosis by bile duct ligation (BDL) and sham-operated controls.
| Intravenous | Oral | |||
|---|---|---|---|---|
| Parameter | Sham | BDL | Sham | BDL |
| AUC (μgh/mL) | 9.33 ± 2.70 | 9.00 ± 1.80 | 27.12 ± 3.72 | 14.11 ± 5.49* |
| Vd (L/kg) | 0.36 ± 0.070 | 0.37 ± 0.09 | ||
| CL (L/hkg) | 0.17 ± 0.04 | 0.16 ± 0.04 | ||
| Half-life (h) | 1.89 ± 0.41 | 2.16 ± 0.39 | ||
| Cmax (μg/mL) | 10.98 ± 3.70 | 5.04 ± 1.25* | ||
| tmax (h) | 0.20 ± 0.02 | 0.62 ± 0.17* | ||
| F (%) | 91 | 48 | ||
Data are presented as mean ± SEM of 6 animals. * Statistically significantly different from sham-operated animals (p< 0.05).
Ketorolac pharmacokinetics were determined in shamoperated rats and in animals submitted to an irreversible obstruction of bile flow. This model is widely used, since it shows several analogies with the condition of cholestasis and cirrhosis in humans.24,25 At the end of the fourweek period of biliary obstruction, rats exhibited an established liver damage, reflected by the presence of acites, by the increase of plasma enzymes activities, and fibrosis. The activity of γ-GTP increased about 14 times, while that of ALT augmented only about twice. This was expected, as γ-GTP is a marker of cholestasis, while ALT is a marker of necrosis.30 We observed a six-fold accumulation of hydroxyproline in the liver of animals with BDL compared to sham-operated controls, indicating an important degree of fibrosis.
Ketorolac pharmacokinetics after intravenous administration did not show any significant alteration in BDL animals with regard to sham-operated controls. These data suggest that BDL-induced cirrhosis does not modify ketorolac distribution and systemic elimination. Nonetheless, liver damage produced a significant reduction in oral ketorolac bioavailability. AUC and Cmax were significantly reduced in about half while tmax was prolonged about three times. These results indicate that BDL reduced both, the extent and rate of ketorolac absorption despite the fact that drug distribution and systemic clearance were not altered. These results were unexpected, as it is frequently assumed that liver damage increases drug bioavailability or produces no change. Increases in bioavailability could be due to a decreased hepatic first-pass effect of by the presence of porto-systemic shunts which allow a drug to bypass parenchymal liver tissue and thus reducing first pass extraction.14 It should be noted, however, that most pharmacokinetic studies showing bioavailability increases in liver damage have been performed for compounds which are cleared, at least partially, by hepatic metabolism and/or excretion. Poorly metabolized drugs with no biliary excretion, such as ketorolac, are seldom characterized.
A reduction in oral drug bioavailability in liver damage can be explained by an impaired drug transfer from the gastrointestinal lumen to the splachnic circulation, by drug transfer to ascitis fluid before arrival to the systemic circulation or by a decreased hepatic first pass effect. In the case of oral ketorolac bioavailability in experimental cirrhosis, an alteration on hepatic first pass effect can be discarded. BDL did not result in a significant change in ketorolac total systemic clearance. Thus, it seems unlikely that hepatic biotransformation could play a significant role in the observed alterations in oral bioavailability. Furthermore, it has been reported that ketorolac is poorly metabolized, being mainly excreted as the unchanged drug.2,16 In agreement with these data, we observed that, in sham-operated animals, ketorolac absolute bioavailability by the oral route was almost complete (F = 90%), indicating a negligible participation of first pass extraction.
The significantly reduced ketorolac absolute bioavailability by the oral route (from 90% to 48%) observed in experimental cirrhosis could be due to an impaired transfer from the gastrointestinal lumen to the splachnic circulation. As mentioned above, BDL rats exhibited an important degree of liver fibrosis. Fibrosis is an important consequence of chronic liver diseases, which consists of deposition of connective tissue around the hepatic sinusoids. As a global consequence of collagen accumulation, liver blood flow resistance ensues, causing portal hypertension which, in turn may lead to stasis in the splachnic circulation and ascites.18,20
Ketorolac transference from the gastrointestinal lumen to the splachnic circulation likely occurs by passive diffusion, since it has been shown that its oral pharmacokinetics are linear in both, experimental animals and humans.31,32 Thus, the transference process should be described according to Fick’s law of diffusion.33 In such case, drug transference depends on the drug concentration gradient established between gastrointestinal lumen and the splachnic circulation. If the drug is efficiently cleared from the capillaries irrigating the stomach and small intestine, capillary blood concentration will be maintained at near-zero values, favouring drug transference from the gastrointestinal lumen. In the case of stasis, drug clearance from the capillaries may be less efficient, thus blood concentration can rise to significant values impairing drug transference from the gastrointestinal lumen. It has been reported that rats with BDL, hepatic circulatory disturbance is caused by narrowing of peripheral portal vein branches and sinusoidal stenosis, with no perceptible change in hepatic vein branches.19 Narrowing of peripheral portal vein branches can lead to a reduced splachnic blood flow affecting ketorolac absorption, without affecting systemic clearance and thus could be involved in the reduced oral ketorolac bioavailability in BDL rats.
Other possible explanation for a reduced oral ketorolac bioavailability is drug transference to ascites fluid, as rats with DBL exhibited ascites. Ascites is a consequence of portal hypertension due to the infiltration of plasma through the walls of capillaries18 and thus it can be argued that ketorolac had been sequestered in the ascites compartment, impairing its transference to the systemic circulation. With the information available at present, it is not possible to establish whether reduced oral ketorolac bioavailability in experimental cirrhosis is the result of impaired drug transfer from the gastrointestinal lumen to the splachnic circulation, or drug being sequestered in ascites fluid or whether both processes are the cause for it. Further research is required to elucidate this issue.
Despite the assumption that drug bioavailability is increased in liver damage, our results show that, at least in experimental BDL-induced cirrhosis, ketorolac bioavailability is significantly reduced when given by the oral route, being unchanged by the intravenous route. The present data demonstrate that liver damage can alter the pharmacokinetics of poorly metabolized compounds, and that these observations can be unexpected considering data from extensively metabolized drugs. Pharmacokinetic changes can also be due to alterations in drug absorption, which have not been studied thoroughly. In fact, Pages and colleagues15 observed a prolonged tmax for oral ketorolac in patients with alcoholic cirrhosis while systemic clearance remained unchanged. These data suggest that in these patients, as in our experimental animals, ketorolac absorption from the gastrointestinal tract was altered despite a lack of significant changes in clearance. In summary, our results demonstrate that liver damage can alter pharmacokinetics even in absence of effects on drug metabolism. The overall effect of liver diseases on drug kinetics appears to be more complex than assumed. Hence, uncritical extrapolation of data should be avoided. Systematic pharmacokinetic studies in different types of liver damage should be performed to fully characterize this matter. The use of experimental models of liver damage, mimicking the different disease states found in the clinical situation, appears to be a suitable strategy for this purpose.
AcknowledgmentsThe authors express their gratitude to Ms. Patricia González for the preparation of the manuscript and figures, and to Ms. Lourdes González, Mr. Mario Gil Moreno, Mr. Ramón Hernández, and Mr. Benjamín Salinas for their excellent technical assistance. This study was supported in part by grant 38940-M from Conacyt, Mexico. Liliana Rivera-Espinosa and Mónica Ordaz-Gallo were fellows of Conacyt.



