




FAP is an autosomal dominant inherited disease, characterized by systemic deposition of amyloid fibrils in various tissues. The purpose of this study is to describe the gross and microscopic findings of the explanted livers for FAP.
10 patients were transplanted for FAP at our institution. Diagnosis was supported by positive familiar history, clinical data and detection of mutated TTR by electrospray ionization mass spectrometry with Val30Met mutation verified by PCR. All the explanted livers were photographed, fixed in formol and processed according to protocol. Later they were examined with HyE, reticulin, PAS diastasa, Masson trichromic, Congo red with polarised light and immunoreactivity against TTR. The gross aspect was normal. We obtained multiple samples representative of the organ and the hepatic hilium. All of the patients presented with deposits of amyloid substance in the lymph nodes and the nerves of the hepatic hilium These deposits were Congo red positive with a greenish birefringence to polarized light Deposits show immunoreactivity with antihuman TTR. Whereas liver transplantation restores hepatic function in patients with cirrhosis, liver transplantation cures the FAP patient of their genetic defect. Domino transplantation is a procedure in which the index patient receives an organ, while the explanted organ is reused for transplantation into another patient. In conclusion, exclusion of hepatic amyloid deposits which can cause functional alterations in the FAP liver is vital; and is important to study the explanted livers of patients with FAP to confirm the results of the scarce published series.
Hospital Dr. Cosme Argerich. Autonomous City of Buenos Aires
Background/aimsSince Corino Andrade first described Familiar amyloid polyneuropathy (FAP) in a group of portuguese families in 1952, many other with similar manifestations have been reported throughout the world.
FAP is an autosomal dominant inherited disease, characterized by systemic deposition of amyloid fibrils in various tissues, resulting in organ dysfunction and ultimately leading to death.1
The component of amyloid fibrils consist of Transthyretin (TTR) formerly known as prealbumin.2 This is a 127 amino acid protein that functions as a transport protein for thyroxin and saturated retinol-binding protein. A mutant, amyloidogenic TTR molecule is produced in patients with FAP as a result of a single amino acid substitution.3 This variant protein is found in plasma and is deposited in the tissues, constituting a biological marker of the disease. It is also detected in patients relatives (asymptomatic carriers) who may even develop the disease and transmit it to their descendants.4
The penetrance of the disease is incomplete, some carriers of the mutant gene are asymptomatics and live past age ninety, others manifest widespread involvement. The gene prevalence is unknown but is considered in at 1 in 100,000 to 1 million.5
The first mutation within the TTR gene causing FAP was described in 1985 and the gene was localized on chromosome 18q11.2-q12.1.6 Since this study approximately 54 point mutations in the TTR gene have been reported, however the most common worldwide defect is a substitution of the of valine by methionine at position 30 (Met 30).
Most TTR is produced by the liver (98%), with minor sites of synthesis (< 2%) in the choroid plexus, the small intestine and the retinal pigment epithelium of the eye.1 However unlike other inborn errors of metabolism, such as defects of the urea cycle and disorders of fatty acid metabolism or Wilson's disease, in FAP the liver itself it does not seem to be damaged. Amyloid deposits may occur, but liver function is not impaired.7
The disease generally begins between ages 25 and 35, and ultimately fatal 7-10 years after onset of symptoms from progressive cardiac dysfunction, emaciation, renal failure or other complications related to autonomic neuropathies.8 Typical findings include initially loss of skin sensibility of hands and feet walking disability, increased motility of the intestine causing diarrhea and malnutrition, orthostatic dysregulation, cardiomiopathy and proteinuria.9-11 Recently has been described a new endemic area in Argentina where it lives a group of portuguese immigrants families.12
The therapeutic approach to the disease has been made with plasmaferesis and extracorporeal immunoadsorption13 but liver transplantation is the only definitive treatment for FAP and was first successfully performed in Sweden in 1990. The rationale for liver transplantation is to eliminate the main source of mutant TTR production, thereby arresting the progression of amyloid deposition. Since 1990, orthotopic liver transplantation has been performed at multiple international transplant centers for FAP.14
There is only few reports about the histopathological findings in necropsy material or liver resection, and the previously reported necropsic cases suggest that endoneural deposits cause peripheral nerve fiber loss due to local ischemia.15 The purpose of this study is to describe the gross and microscopic findings of the explanted livers for TTR FAP.
Material and methodsBetween January 1997 and September 2002, 10 patients were transplanted for FAP at our institution where constitutes 5.26% of liver transplantation cases.16 Five of them belonged to the same family (two siblings on one hand, two siblings for other and a cousin). Two patients belong to another family (two siblings).
Diagnosis was supported by positive familiar history, clinical data and detection of mutated TTR by electrospray ionization mass spectrometry with Val30Met mutation verified by PCR. Mean ages were: at onset of symptoms 28.75 (SD 3.1) and at transplantation 35,79 (SD 4,8). Average polyneuropathy disability score was II. Autonomic dysfunction was manifested by gastrointestinal symptoms: diarrhea 50%, diarrhea and constipation 20%, faecal incontinence 50%, sexual impotence 70%; cardiovascular symptoms: denervation by heart rate variability 80%, heart conduction disturbances 30% and orthostatic hypotension 50%. Pretransplant modified body mass index was 726,38 (SD: 143,48).
All the explanted livers were photographed, fixed in formaline and processed according to protocol. Later they were examined with HyE, reticulina, PAS diastasa, Masson trichromic and Congo red. The same ones were examined with polarised light. With this same technique were studied the elements of the hepatic hilio and cellular subcutaneous tissue from surgical procedures.
Immunomarcation against TTR was performed in available tissue.
ResultsThe gross aspect was normal, the weight mean was of 1,028 g (900 at 1,095 g). The liver explanted from the 10 patients were studied in exactly the same way. We obtained multiple samples representative of each segment of the organ together with another samples from the hepatic hilium to obtain nerve bundles of medium caliber. The samples were studied with all the techniques previously described.
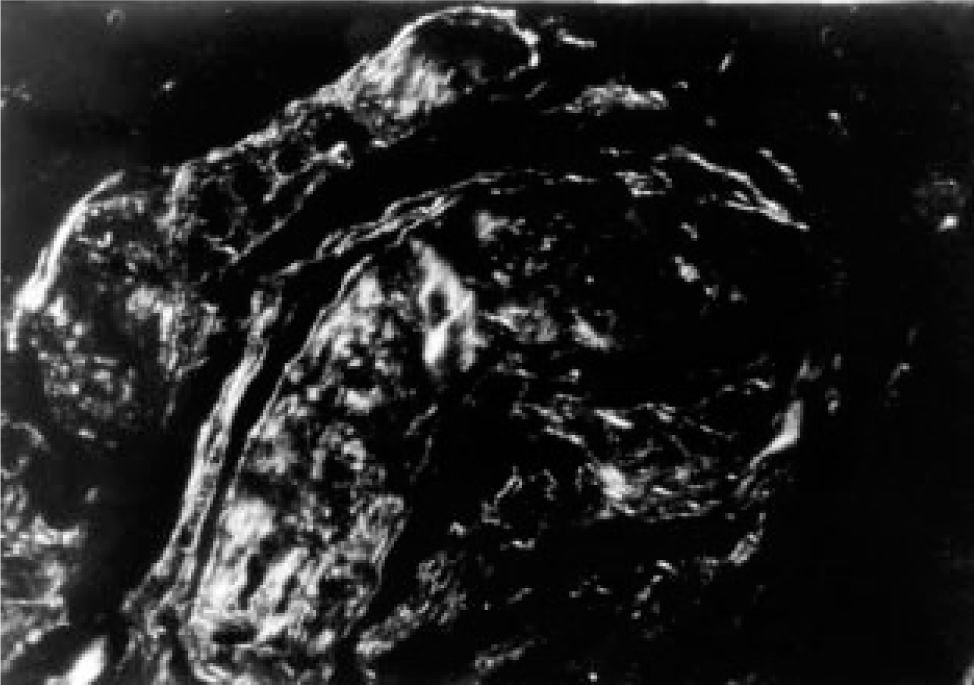
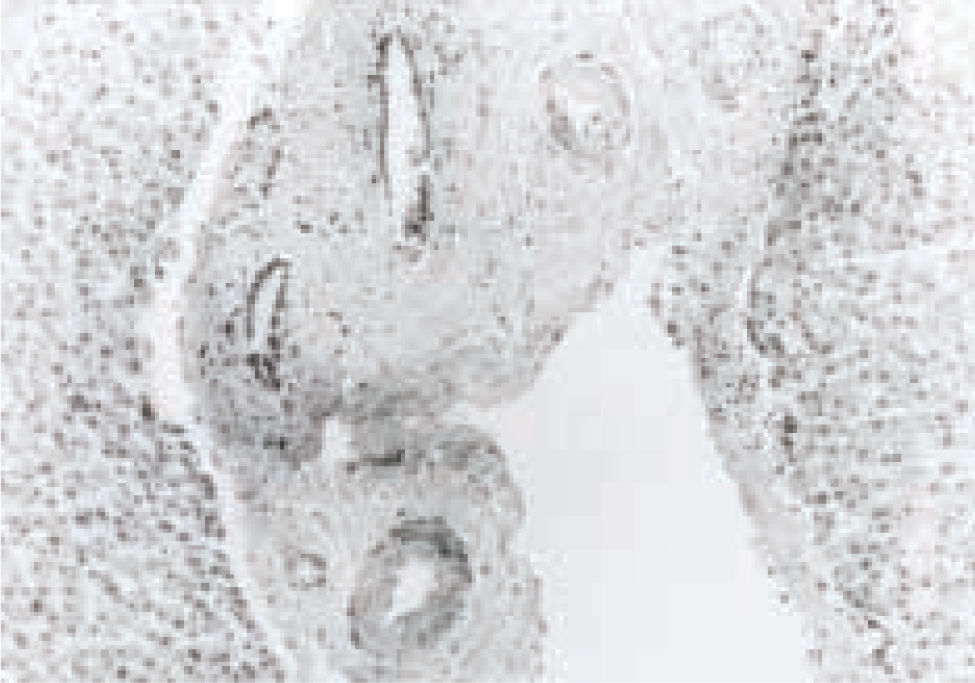
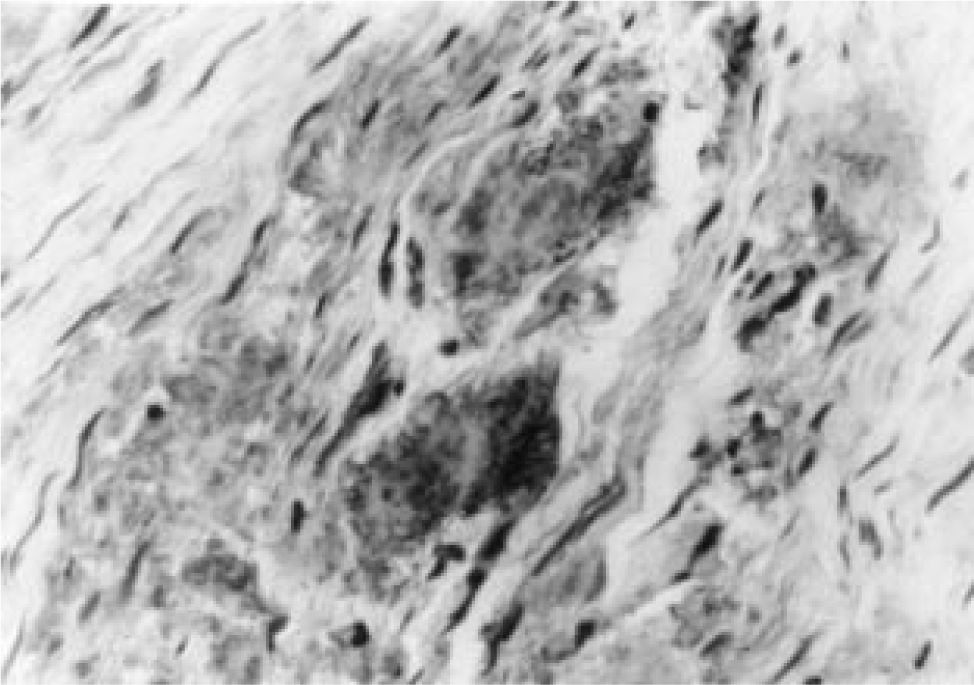
None of the patients revealed significant alterations in the macroscopic study of the liver and nor were there significant structural alterations microscopically in any of the patients. All of the patients presented deposits of amyloid substance in the lymph nodes and the medium caliber nerves of the hepatic hilium (Figure 1), and in those observed in the occasional thick intrahepatic septum. These deposits were Congo red positive with a greenish birefringence to polarised light (Figure 2). Deposits of amyloid were also observed with the same staining characteristics as in the nerve, mostly in arterioles of the portal tracts, which were irregularly enlarged (Figure 3).
.")
.")
.")
Deposits show immunoreactivity with antihuman TTR and TTR was also detected in a minimal proportion in the liver cells (Figure 4).
Discussion.")
In post mortem examination amyloid deposits has been reported in cornea, heart, thyroid, suckles, kidneys, adrenal glands, gastrointestinal tract, pancreas and bladder, as well as in the vessels of lungs, liver, spleen, lymph nodes, bone marrow, uterus, ovary, fallopian tubes and around skin glands.17
In our cases the affectation but important it is perivascular and endoneural.
In the liver, amyloid deposits occurs in the walls of small arteries in Glisson’s sheaths but not in the walls of the portal, hepatic or central veins; in the sinusoid; around the bile ducts; and around the extrahepatic biliary tract.18
Some authors have proposed ischaemia as a possible cause of nerve fibers loss. The amiloyds deposits cause endothelial proliferation and alter the vascular permeability resulting in oedema and later ischaemia but has not been exactly established the definitive pathogenic mechanism of the nerve lesions.16,17
Liver transplantation is a therapeutic option when the abnormal synthesized protein which is causing a life threatening disease, is primarily synthesized within the liver.6 The improvement in surgical techniques, the new immunosuppressive drugs, and the better handling of complications after transplantation have resulted in increasing number of liver transplantations in patients with genetic liver associated diseases.19 Some of these genetic defects originate within the liver, causing extrahepatic complications, such as LDL receptor deficiency (familiar hypercholesterolemia) oxalosis, and factor VIII/IX deficiency.20 However, the liver as the primary site of synthesis is not affected in these patients. and hepatic failure does not occur. FAP also belongs to this group of diseases.
Whereas liver transplantation restores hepatic function in patients with infectious or toxic induced cirrhosis, liver transplantation cures the FAP patient of their genetic defect, arrests further deposition of mutant proteins in tissues, and result in significant palliation of many of the underlying symptoms.3-21
Domino transplantation is a procedure in which the index patient receives an organ, while the organ of the index patient is reused for transplantation into another patient. Since the onset of extrahepatic complications is in adulthood, FAP can be used as a candidate disease for domino liver transplantation.3
In this setting exclusion of hepatic amyloid deposits which can cause functional alterations in the FAP liver is vital.22
In conclusion, our results suggest that while we await less aggressive solutions, a liver transplant may be useful in the treatment of certain cases of familial amyloidotic polyneuropathy to stop the neurological deterioration of the patients and to avoid the fatal end of the disease.23
We believe that is extremely important to study the explanted livers of patients with FAP to confirm the results of the scarce published series and to establish a domino transplantation program in our and other institutions where it finds cases of patient with this disease.
AcknowledgmentWe thank Dr. Gregorio Chejfec from VA Hines Hospital. Hines, ILL, for the immunohistochemical techniques against TTR carried out in our first six cases.



