





Renal dysfunction is a common finding in cirrhotic patients and has a great physiologic, and therefore, prognostic relevance. The combination of liver disease and renal dysfunction can occur as a result of systemic conditions that affect both the liver and the kidney, although primary disorders of the liver complicated by renal dysfunction are much more common. As most of the renal dysfunction scenarios in cirrhotic patients correspond to either prerenal azotemia or hepatorenal syndrome (HRS), physicians tend to conceive renal dysfunction in cirrhotic patients as mainly HRS. However, there are many systemic conditions that may cause both a “baseline” chronic kidney damage and a superimposed kidney dysfunction when this systemic condition worsens. The main aim of this article is to review some of the most important non prerenal non-HRS considerations regarding acute on chronic kidney dysfunction in cirrhotic patients, including renal manifestation of related to non-alcoholic steatohepatitis (NASH) viral hepatitis, the effect of cardiorenal syndrome in cirrhotics and corticosteroid-deficiency associated renal dysfunction.
Renal dysfunction is a common finding in cirrhotic patients and has a great physiologic, and prognostic relevance. The combination of liver disease and renal dysfunction can occur as a result of systemic conditions that affect both the liver and the kidney, although primary disorders of the liver complicated by renal dysfunction are much more common.
Even when renal manifestations of nonalcoholic steatohepatitis (NASH) viral hepatitis, cardiovascular disease and corticosteroid deficiency in cirrhotic patients are well documented causes of both chronic and acute kidney damage, rarely they are evaluated during the diagnostic-therapeutic approach of renal damage on cirrhotic patients. In the following sections, a concise review of renal manifestation of NASH, viral hepatitis, cardiorenal syndrome and corticosteroid deficiency on cirrhotic patients will be made.
2NASHAmong patients with nonalcoholic fatty liver disease (NAFLD), 28% have impaired renal function, 5% abnormal GFR, 18% significant proteinuria and 5% significant proteinuria and abnormal GFR. Type 2 diabetes mellitus, raised hepatic transaminases and advanced fibrosis have been described as independent predictors of impaired renal function [1]. A growing body of data suggests liver–kidney crosstalk in NAFLD, including [2]:
- 1.
Altered activation of angiotensin converting enzyme (ACE)-2: The Renin–Angiotensin System (RAS) plays a key role in the pathogenesis of obesity-related disorders, including NAFLD and CKD. Adipocytes express all RAS components. The kidney and liver also express all RAS components, and experimental studies support a role for both systemic and local (renal and hepatic) paracrine/autocrine ACE-Angiotensin II-type 1 activation in the pathogenesis of NAFLD and CKD [2].Renin–angiotensin II type 1 receptor axis activation is linked to hepatic steatosis, insulin resistance, hepatic inflammation and fibrosis, as well as efferent artery vasoconstriction, glomerular hypertension and endothelial cell injury, giving place to glomerular hyperfiltration, hypertrophy and sclerosis, as well as tubulointerstitial inflammation and fibrosis [3].In the Fatty Liver Protection Trial by Telmisartan or Losartan Study (FANTASY), telmisartan improved also necroinflammation, NAFLD activity score, and fibrosis stage in NASH, as well as microalbuminuria [4].
- 2.
AMP-activated kinase: 5′-AMP-Activated Protein Kinase is a ubiquitous heterotrimeric serine/threonine kinase, that functions as a cellular energy sensor and a key regulator of cellular metabolism [5]. AMPK is activated under conditions of calorie restriction or high energy demand that deplete cellular ATP stores and increase the AMP/ATP ratio, whereas it is inhibited under conditions of calorie excess such as obesity. Activation of this kinase is associated with diminished hepatic inflamation and fibrogenesis, as well as endotelial protection and diminished toxic lipid deposition in renal endothelium, mesangial cells, podocytes and tubule cells [5].
- 3.
Nutrient/energy sensor sirtuin-1: Sirtuins (SIRTs) are a group of seven NAD+-dependent histone/protein deacetylases belonging to the silent information regulator-2 family. The deacetylation of proteins and histones results in an up- or downregulation of gene transcription and protein function [6].Upon calorie restriction, increased intracellular NAD+concentrations activate SIRT1 to promote chromatin silencing and transcriptional repression eventually resulting in amelioration of glucose and lipid homeostasis in the liver, muscle, and adipose tissue; furthermore, SIRT-1 activation also showed anti-oxidant effects, downregulates proinflammatory genes in the liver and adipose tissue [7], and promotes autophagy [8]. SIRT-1 activation ameliorated also experimental CKD through indirect effects, mediated by improved glucose and lipid metabolism, and direct, renal-specific effects: SIRT-1 preserves podocyte function and glomerular barrier integrity by downregulating the tight junction protein claudin-1 [9]; it also downregulates proinflammatory and pro-fibrogenic pathways [10].
- 4.
Lipoprotein dysmetabolism: NASH and CKD are characterized by free cholesterol accumulation in hepatic and renal cells, which triggers oxidative stress inflammation and fibrosis in the liver and kidney [11]. Liver fat accumulation induces atherogenic dyslipidemia through oversecretion of large VLDL particles, triggering intravascular lipoprotein remodeling, and the typical atherogenic lipoprotein phenotype of reduced small high-density lipoprotein particles, and increased circulating levels of small, dense LDL and oxidized LDL particles, which bind to receptors on glomerular endothelial cells, mesangial cells, tubular cells, and interstitial macrophages, and trigger glomerular injury, mesangial cell proliferation, and foam cell formation [12,13].The master nuclear regulators of cellular cholesterol metabolism in the liver and kidney are the nuclear transcription factors Sterol regulatory element-binding proteins (SREBP-2) and the famesoid X receptor (FXR), expressed in all hepatic cell lineages, glomerular and proximal tubular cells [12,14].Evidence from NASH patients and from experimental diet-induced models of NASH and CKD indicate inappropriate SREBP-2 upregulation, with consequent increase in cholesterol synthesis, influx, and retention, as well as accumulation of intracellular toxic free cholesterol, impaired bile acid synthesis, and reduced bile acid-stimulated FXR activation: the resulting intracellular cholesterol overload triggers multiple molecular pathways leading to cell death, inflammation, and fibrosis in the liver and kidney [2,14,15].FXR activation by potent, semisynthetic bile acids such as obeticholic acid improved NAFLD activity score and fibrosis in the multicenter, double-blind, randomized ‘FXR Ligand NASH Treatment (FLINT)’ trial in NASH [16] and reversed renal lipid accumulation, oxidative stress, inflammation, fibrosis, and proteinuria in diet-induced experimental CKD [14].
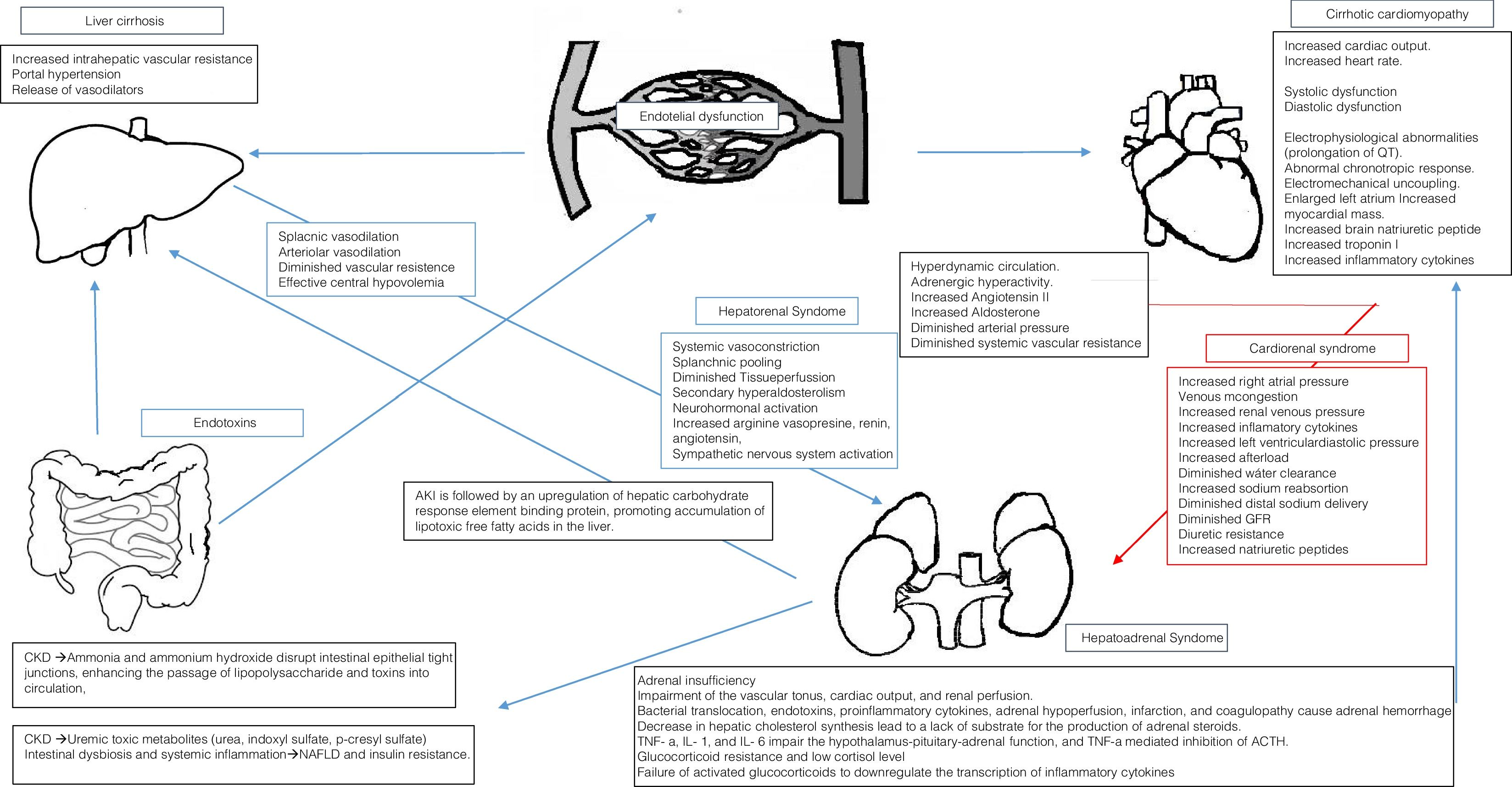
AKI is followed by an upregulation of hepatic carbohydrate response element binding protein-mediated de novo lipogenesis, and by downregulation of PPAR−/-mediated free fatty acids oxidation and diglyceride acyltransferase – mediated Free fatty acids incorporation into triglycerides, thereby promoting accumulation of lipotoxic free fatty acids in the liver [17].
CKD induces intestinal dysbiosis and systemic inflammation, promoting NAFLD and insulin resistance. Mechanisms include the accumulation of uremic toxic metabolites that are normally eliminated by the kidneys, including urea, indoxyl sulfate, p-cresyl sulfate (p-CS) and trimethylamine-N-oxide [5,18].
The accumulation of urea may lead to influx into the gastrointestinal lumen, where it is hydrolyzed to ammonia by microbial urease, then converted to ammonium hydroxide. Ammonia and ammonium hydroxide disrupt intestinal epithelial tight junctions, enhancing the passage of lipopolysaccharide and other toxic luminal compounds into the circulation, with alteration of intraluminal pH and promotion of the growth of urea-metabolizing microbial strains at the expense of carbohydrate-fermenting strains [18].
3Renal complications of viral hepatitisRenal impairment in viral hepatitis involves the formation of immune-complexes with the virus, antibody production against altered hepatocytes or other infected cells, or direct virus cytopathic effects [19] (Table 1) summarizes of the main viral hepatitis associated renal disease.
Viral hepatitis related renal disease. Renal impairment in viral hepatitis involves the formation of immune-complexes with the virus, antibody production against altered hepatocytes or other infected cells, or direct virus cytopathic effects.
| Hepatitis virus | Related kidney disease | Disease-specific characteristics | Ref. |
|---|---|---|---|
| HAV | AKI | Due to dehydration and/or immune complex deposition | [24,26] |
| Interstitial nephritis | Rarely seen | [24,26] | |
| Mesangio proliferative glomerulonephritis. | Rarely seen | [24,26,71] | |
| IgA nephropaty | Most frequent glomerulonephritis associated with HAV infection | [24,26] | |
| HBV | MN | − Associated with chronic carrier, HbeAg IgG− Likely to present with microscopic hematuria and lower complement levels.− Presence of mesangial or subendothelial immune deposits and the typical subepithelial localization, may be a clue to suggest secondarymembranous nephropathy.− Phospholipase A2 receptor has been implicated as the major antigen involved− Deposition of hepatitis B e antigen (HBeAg) and hepatitis B e antibody (anti-HBe) immune complexes in the subepithelial region of the glomerular basement membrane is pivotal.More common in children and may resolve spontaneously, usually in association with seroconversion from HBeAg to anti-HBe.− Spontaneous resolution is relatively uncommon in adults, some patients show progressive renal deterioration. | [19,28–30][23,32,72,73,77,78] |
| MPGN | − Characterized by deposition of circulating antigen-antibody complexes in the mesangium and subendothelial space.− HBsAg and HBeAg deposition implicated.− HBV is a rare cause of mixed cryoglobulinemia, which can be associated with MPGN:− Patients present with nephrotic-range proteinuria, microscopic hematuria, hypocomplementemia and positive for rheumatoid factor.− 75% have impaired renal function and 50% of patients dies or developed ESRD despite immunosuppressive and antiviral treatments. | [19,28–30][23,32,77,78] | |
| PAN | − Clinical features of HBV-associated PAN are similar to idiopathic PAN.− Typically occurs within four months after the onset of HBV infection.− Suggested by the findings of HBsAg, HBeAg, and HBV DNAin the serum. Deposition of circulating antigen-antibody immune complexes in the vessel wall triggers downstream inflammatory processes. | [23,28,55,74,79] | |
| HCV | Mixed cryoglobulinemia syndrome | − Clinical manifestations include hematuria, proteinuria (which may be in the nephrotic range), and renal impairment. Laboratory studies indicate hypocomplementemia and circulating cryoglobulins.− The complement components, C3, C4, and C1q, are usually low; reductions in C4 are usually greater than reductions in C3.− Cryoglobulins are most commonly type II, characterized by a polyclonal immunoglobulin G (IgG) and monoclonal immunoglobulin M (IgM) kappa rheumatoid factor directed against the Fc portion of IgG.− HCV-containing immune complexes are directly involved in the pathogenesis of disease. HCV RNA and HCV IgG antigen-antibody complexes are highly concentrated in the cryoprecipitate.− HCV antigens have been detected along the glomerular capillary walls and in the mesangium of patients with HCV-related glomerulonephritis, mainly in those with MPGN and positive HCV RNA.− The glomerular basement membrane shows double contours caused by the interposition of monocytes between the basement membrane and the endothelium− Subendothelial deposits of IgM, IgG, and complement components are observed by immunofluorescence and electron microscopy. Vasculitis of small renal arteries is sometimes present. | [28,75-78] |
| Membranous nephropathy | − Several small studies have suggested that membranous nephropathy may be induced by chronic HCV infection, although the data are conflicting.− Membranous nephropathy is reported to be higher in HCV-positive compared with HCV-negative renal transplant patients.− Complement levels tend to be normal, and neither cryoglobulins nor rheumatoid factor is present. | [75,76] | |
| PAN | − PAN is reported to occur at a mean of two years after the diagnosis of HCV infection.− The patients may have typical symptoms of PAN and necrotizing inflammation of medium or small arteries in biopsy specimens; 26% of tested patients are reported to be also positive for hepatitis.− Patients infected with HCV who develop PAN may have more severe systemic manifestations of vasculitis compared with those HCV patients who develop mixed cryoglobulinemia vasculitis.− Compared with HCV-related mixed cryoglobulinemia patients more commonly had fever, weight loss, hypertension, gastrointestinal tract involvement, severe mononeuropathy, and elevated C-reactive protein. | [78,79] | |
| HEV | MPGN, IgA nephropathy.Nephroangiosclerosis.Membranous glomerulonephritis. | − Almost exclusive of genotype 1 and 3. | [80] |
AKI is frequent in patients that have fulminant hepatitis A and is found in 1.5–4.7% of patients with non-fulminant hepatitis A. The most frequent glomerulonephritis associated with HAV infection is IgA nephropathy, which sometimes presents with nephrotic syndrome and can spontaneously resolve [20–22].
HBV may be associated with a variety of renal diseases, being the most common: Membranous nephropathy, Membranoproliferative glomerulonephritis (MPGN) and Polyarteritis nodosa (PAN) (Table 1) [19,23–25].
Renal disease associated with HBV infection most commonly occurs in endemic areas where infection is more likely to occur during infancy and early childhood, increasing the probability of becoming a chronic carrier [26]. HBV vaccination has decreased the incidence of HBV-related membranous nephropathy and MPGN [26]. Some patients with HBV-related renal disease have a history of active hepatitis, but a large proportion have only mild to moderate elevations in serum aminotransferases [27].
The pathogenetic role of HBV infection has been documented primarily by the demonstration of hepatitis B antigen-antibody complexes in the renal lesions via immunofluorescence microscopy, including deposition of HBeAg in membranous nephropathy. The relationship between HBV variants (e.g., precore and core promoter mutations) and renal disease has not been well described. HBV DNA and HBV RNA have been localized to glomerular and tubular cells in affected patients; however, the role of these viral nucleic acids in the development of renal injury remains to be confirmed [28,29].
Purified HBV can induce human glomerular mesangial cell proliferation and expression of type IV collagen. In addition, HBV X protein has been shown to induce a proinflammatory phenotype in renal tubular epithelial cells, although the impact of increased tubulointerstitial inflammation on glomerular pathology has not been investigated [30,31].
The treatment of HBV-associated renal diseases consists of antiviral therapy. Immunosuppressive therapy, with or without plasmapheresis, is used only in select patients, e.g. rapidly progressive glomerulonephritis (RPGN) or PAN with severe manifestations and, when it is administered, it should be used in combination with antiviral therapy [27].
There is a strong and likely causal association between chronic HCV infection and glomerular disease. The major glomerular diseases associated with HCV infection include Mixed cryoglobulinemia syndrome, membranous nephropathy and PAN. Crescentic glomerulonephritis may be superimposed on any of these glomerular lesions [32,33].
Less common lesions have been reported in HCV-infected patients, including FSGS, proliferative glomerulonephritis, and fibrillary and immunotactoid glomerulopathies. Clinically silent glomerular disease has been described primarily in patients who have undergone liver transplantation for cirrhosis due to chronic HCV infection [32,33].
Chronic HCV infection is associated with an increased risk of developing end-stage renal disease (ESRD). Elevated serum levels of HCV RNA (>167,000IU/mL) and HCV genotype 1 are strong predictors of ESRD. Glomerular diseases associated with HCV can also occur in renal allografts [32].
Patients with severe and progressive HCV-associated glomerulonephritis should undergo antiviral treatment. Most patients with less severe glomerular disease associated with HCV should also have antiviral therapy provided they do not have decompensated cirrhosis. Patients with severe and progressive mixed cryoglobulinemia and PAN are also treated with immunosuppressive drugs [32,33].
HEV infection can lead to acute and chronic hepatitis as well as to extrahepatic manifestations [34]. The mechanism of HEV-induced kidney disease could be immune-driven in a manner similar to that with HCV [35]. Ribavirin can be used to obtain a rapid viral clearance. Antiviral administration has reported to led to complete recovery in most patients, whereas end-stage renal disease has also been reported [35] (Table 1).
4Cardiorenal syndrome in cirrhoticsIn 1953, Kowalski and Abelmann [36] showed the existence of a circulatory dysfunction specific to liver cirrhosis. Nowadays, “Cirrhotic cardiomyopathy” (CCM) defines a specific form of cardiac dysfunction characterized by blunted contractile responsiveness to stress stimuli and altered diastolic relaxation with electrophysiological abnormalities, in the absence of a known cardiac disease [37,38].
Proposal of diagnostic criteria for cirrhotic cardiomyopathy agreed upon at the 2005 World Congress of Gastroenterology in Montreal [39] (Table 2). It has been proposed to improve these criteria by including the dysfunction of the right ventricle as an important parameter of cardiac damage caused by cirrhosis [40]. CCM patients also have biventricular diastolic dysfunction at rest, larger left and right atria, higher systolic pulmonary arterial pressure and left ventricular mass [41].
Proposal of diagnostic criteria for cirrhotic cardiomyopathy agreed upon at the 2005 World Congress of Gastroenterology in Montreal. CCM defines a specific form of cardiac dysfunction characterized by blunted contractile responsiveness to stress stimuli and altered diastolic relaxation with electrophysiological abnormalities, in the absence of a known cardiac disease [38,39].
| Systolic dysfunction | Resting ejection fraction<55%Blunted increase in cardiac output with exercise or pharmacological stimuli |
| Diastolic dysfunction | Early diastolic atrial filling ratio (E/A ratio)<01.0 (age corrected)Deceleration time (DT)>200msProlonged isovolumetric relaxation time>80ms |
| Supportive criteria | Electrophysiological abnormalities (prolongation of QT).Abnormal chronotropic response.Electromechanical uncoupling.Enlarged left atrium Increased myocardial mass.Increased brain natriuretic peptide and pro-peptideIncreased troponin I |
Portal hypertension leads to a state of peripheral vasodilatation due to a release of vasodilator mediators such as nitric oxide, carbon monoxide, and prostacyclin. Progressive hepatic decompensation results in blood volume redistribution, with increased splanchnic blood flow and relatively decreased central circulation. The baroreceptors may be deactivated to compensate for the reduction in the central and arterial blood flow, with increased sympathetic nervous activity [38,42].
Hyperdynamic circulation in cirrhosis is an appropriate initial response to the splanchnic arterial vasodilation. Patients with advanced cirrhosis exhibit also with increased cardiac output. This hyperdynamic state arises from a marked splanchnic arterial vasodilation and reduced systemic vascular resistance. Increase in vasodilation due to portosystemic shunting and bacterial translocation increase the hyperdynamic circulation and central hypovolemia [43,44].
At rest, the intra-cardiac pressures are normal because the reduced afterload secondary to the systemic vasodilation compensates for both preload reduction and contractile dysfunction. This circulatory state, which resembles those of high cardiac output, results from increased blood flow and is defined as heart failure due to volume overload [43,44].
Although the cardiac output is increased in patients with advanced cirrhosis, this increase is insufficient to maintain an adequate arterial blood pressure and renal perfusion and hence to prevent renal vasoconstriction and other counter-regulatory mechanisms. The recognition and understanding of a cardiac involvement would change the focus in the prevention and treatment of renal dysfunction in cirrhosis and in the development of new treatments [45].
Renal failure and decreased effective blood volume in cirrhosis, is not only a consequence of arterial vasodilatation but also of impaired cardiac contractility suggesting the existence of a cardiorenal syndrome in cirrhosis. It has been hypothesized that the cardiorenal relationship in decompensated cirrhosis is a result of an acute stress superimposed on an abnormal circulatory state. There may therefore be a link between the acute cardiac dysfunction and the development of type 1 HRS [46,47].
Cardiac inotropic and chronotropic dysfunction is a key component of the circulatory dysfunction observed in HRS. Maladaptive neurohormonal activation represents the key pathophysiologic mechanism linking cardiac dysfunction to HRS. It is also the cornerstone of the pathways involved in the development of cardiorenal syndrome [48].
In patients with cardiac dysfunction, a number of factors, such as low cardiac output and use of diuretics, can lead to elevated levels of neurohormones and their downstream renal adverse consequences, such as reduction in glomerular filtration rate and impaired sodium and water excretion (low forward flow) [49]. Increased cardiac right-sided pressures could result in renal venous congestion and further deterioration in renal hemodynamics and function (high backward pressure) [50,51]. As such, cardiac dysfunction could contribute to further deterioration of renal function in HRS, whereby there is already a tendency for activation of the neurohormonal axis [50,51].
The treatment of CCM is directed to left ventricular failure, with sodium restriction, diuretics, and after load reduction. Liver transplantation may improve or normalize cardiac function. Non-selective beta-blockers have shown to reduce the prolonged QT interval and might reduce the hyperdynamic load in patients with cirrhosis, but whether the correction of the QT interval has a positive effect on prognosis is doubtful [52]. In patients with refractory ascites, the use of beta-blocker has a risk of increased mortality [53]. In a revision of 10 hemodynamic studies on liver cirrhosis, Tripathi and Hayes [54] concluded that carvedilol is a potent agent to reduce portal pressure. However, careful dosing is necessary to minimize adverse effects, especially reduction in the mean arterial pressure [54].
Angiotensin converting enzyme inhibitors can improve diastolic function in patients with cirrhosis by decreasing the ventricular thickness and dilation, but they should be used in the early phases of cirrhosis because of the risk of hypotension and hepatic-renal syndrome in later phases [55]. In an animal model of bile duct ligation induced cirrosis in male rats, chronotropic responses increased after four weeks treatment with losartan, but it was not significant. The pathological study showed that losartan could not decrease fibrosis in atria in losartan treated cirrhotic group. These results might be considered as angiotensin II role in cirrhotic cardiomyopathy, but further studies are required to elaborate the mechanism as well as the possible advantage of losartan [56]. Even though these drugs reduce the portal pressure in patients with cirrhosis, they should be discontinued if ascites appears, as they can further aggravate the systemic vasodilation state and increase the risk of hepatorenal syndrome [57,58].
Liver transplant is the only effective treatment established for patients with final stage liver disease associated with CCM. Liver transplantation has been shown to reverse the systolic and diastolic dysfunction and prolonged QT interval [59].
5Corticosteroid deficiencyRelative adrenal insufficiency (RAI) is frequently observed in patients with cirrhosis. In a prospective study evaluating adrenal function in a population of non hospitalized stable cirrhotic patients; 15.23% had evidence of adrenal insufficiency. These patients were not statistically different from those with normal adrenal function in levels of serum creatinine or bilirubin, MELD score, or presence of cirrhosis related complications. Significant differences were seen in mean international normalized ratio and serum sodium. Patients with a sodium level<135mEq/L had a higher rate (31.25%) of adrenal insufficiency, suggest a possible role for adrenal dysfunction as a contributing factor in hyponatremia in cirrhosis independent of other known factors of neurohormonal activation secondary to systemic vasodilation [60].
In a systematic review and meta-analysis using the Ovid-MEDLINE, EMBASE, and Cochrane Library databases where 16 studies were included, the prevalence of RAI was 49.4%, and cirrhotic patients with acute critical illnesses such as sepsis were more likely to have RAI compared to those without critical illnesses [61]. Patients with RAI had poorer survival rates and an increased risk of complications such as bleeding and HRS compared to those without RAI. Corticosteroid therapy had a beneficial effect on critically ill cirrhotic patients in terms of hospital survival rate [61].
Among patients with acute gastroesophageal variceal bleeding, critical illness-related corticosteroid insufficiency occurred in 29.9%. The patients with critical illness-related corticosteroid insufficiency had higher rates of treatment failure and 6-week mortality. The cortisol response to corticotropin was inversely correlated with Model for End-Stage Liver Disease and Child–Pugh scores and positively correlated with the levels of high-density lipoprotein and total cholesterol. Hypovolemic shock, high-density lipoprotein, platelet count, and bacterial infection at inclusion are independent factors predicting critical illness-related corticosteroid insufficiency [62].
In patients with cirrhosis, severe sepsis and septic shock, it has been reported that mean arterial pressure, serum bilirubin, vasopressor dependency, and bacteriemia were independent factors that predicted adrenal insufficiency. In patients with cirrhosis, severe sepsis and septic shock, RAI is related to functional liver reserve and disease severity and is associated with hemodynamic instability, renal dysfunction, and increased mortality [63].
It is known that insufficient adrenal cortisol response causes further impairment of the vascular tonus, cardiac output, and renal perfusion. Recent research indicates that development of hepatic nephropathy represents a continuous spectrum of functional and structural dysfunction and may be precipitated by the inherent immunologic, adrenal, and hemodynamic incompetence in cirrhosis [64,65].
RAI-associated hemodynamic instability, renal dysfunction, and increased mortality was first described in critically ill patients with sepsis, but recent studies suggest that it may also be present in patients with cirrhosis without sepsis because of the similar hemodynamic and immunologic disturbances (i.e. hyperdynamic circulation, hypoperfusion, increase in lactate and liver enzymes) [66,67].
Bacterial translocation, endotoxins, proinflammatory cytokines, adrenal hypoperfusion, infarction, and coagulopathy that cause adrenal hemorrhage may depress the adrenal synthesis of steroids or the condition may be a result of glucocorticoid resistance. RAI seems to be related to the functional liver reserve and disease severity and has been referred to as the “hepatoadrenal syndrome”. The liver is the primary site of cholesterol synthesis, and a decrease in cholesterol has been shown in cirrhotic patients, which may lead to a lack of substrate for the production of steroids in the adrenal gland [66,67].
TNF-a, IL-1, and IL-6 have been shown to impair the hypothalamus-pituitary-adrenal function, and TNF-a mediated inhibition of ACTH. The glucocorticoid resistance and low cortisol level in RAI leads to a failure of activated glucocorticoids to downregulate the transcription of inflammatory cytokines in cirrhosis, causing an aggravation of the immunologic incompetence. Low level or insensitivity to cortisol impairs the sensitivity to vasoconstrictors and aggravates the hemodynamic imbalance and thus renal dysfunction and HRS. Furthermore, the existence of an “adrenal insufficiency associated cardiomyopathy” has recently been hypothesized, suggesting that the low glucocorticoid level causes a reduced number and downregulation of beta-adrenergic receptors and cardio-toxic fibrosis of the heart, resulting in impaired cardiac contractility and hence worsening of the circulatory and renal dysfunction that causes HRS [63,67,68].
The effects of corticosteroid therapy in sepsis, severe sepsis and septic shock in cirrhotic patients remain, controversial. Harry [69], in a retrospective study including 20 patients with acute or acute on chronic liver failure documented that the use of 300mg of hydrocortisone per day was associated with a reduction in vasopressor doses, higher incidence of infection and no survival benefit.
In non randomized controlled trial, Marik [70] reported that Hydrocortisone, 300mg/day was associated with reduction dose in norepinephrine an lower mortality rate. However, in a randomized controlled trial involving 39 cirrhotic patients with septic shock, the use of hydrocortisone was associated with a reduction in vasopressor doses and higher rates of shock reversal, but no benefit in 28 day mortality.
6ConclusionsBeside HRS and prerenal AKI, cirrhotic patients are susceptible to a number of conditions that predispose to renal function deterioration. NASH pathogenesis include a wide variety of proinflammatory mechanism involving both the kidney and the liver, including altered activation of angiotensin converting enzyme (ACE)-2 [2]: AMP-activated kinase, Nutrient/energy sensor sirtuin-1 inflammatory and pro-fibrogenic pathways [10]. Lipoprotein dysmetabolism and altered gut microbiota (Fig. 1).

Renal impairment in viral hepatitis involves various mechanisms: the formation of immune complexes with the virus, antibody production against altered hepatocytes or other infected cells, or direct virus cytopathic effects.
The combination of renal failure and decreased effective blood volume in cirrhosis, is not only a consequence of arterial vasodilation but also of impaired cardiac contractility suggesting the existence of a cardiorenal syndrome in cirrhosis.
RAI is frequently observed in patients with cirrhosis. The causative mechanisms includes bacterial translocation, endotoxins, proinflammatory cytokines, adrenal hypoperfusion, infarction, and coagulopathy that cause adrenal hemorrhage may depress the adrenal synthesis of steroids or the condition may be a result of glucocorticoid resistance. Moreover, RAI seems to be related to the functional liver reserve and disease severity and has been referred to as the “hepatoadrenal syndrome”.
Having a deep knowledge of all of the possible causative mechanisms of renal dysfunction in cirrhotics is mandatory to the clinician to approach acute, chronic or acute-on chronic renal dysfunction in cirrhotic patients with or without hepatorenal syndrome.AbbreviationsACE angiotensin converting enzyme acute on chronic liver failure acute kidney injury hepatitis B core antibody hepatitis B e antibody cirrhotic cardiomyopathy end stage renal disease focal segmental glomerulosclerosis the famesoid X receptor glomerular filtration rate hepatitis A virus hepatitis B core antigen hepatitis B e antigen hepatitis B surface antigen hepatitis B virus hepatitis C virus hepatitis E virus hepatorenal syndrome inmunoglobulin A inmunoglobulin G inmunoglobulin M Kidney Disease Improving Global Outcomes membranous nephropathy membranoproliferative glomerulonephritis non alcoholic fatty liver disease nonalcoholic steatohepatitis polyarteritis nodosa relative adrenal insufficiency renin–angiotensin system rapidly progressive glomerulonephritis sterol regulatory element-binding proteins nutrient/energy sensor sirtuin-1: Sirtuins
The authors have no conflicts of interest to declare.



