



Nonalcoholic-fatty-liver disease (NAFLD) is considered the hepatic manifestation of metabolic syndrome (MetS). Mineralocorticoid receptor (MR) activation is associated with increased risk of MetS but few studies have assessed the role of liver MR on NAFLD. We aimed to evaluate the effect of MR modulation by sodium intake in liver injury in experimental models of NAFLD.
Materials and MethodsC57BL/6J mice were fed either a high-fat-diet (HFD) or a choline/methionine deficient (MCD) diet with different sodium concentrations. Hepatic concentration of lipid species, serum aldosterone levels, expression of MR, proinflammatory and profibrotic markers and liver histology were assessed.
ResultsMice fed with High-Na+/HFD showed a lower MR expression in liver (p = 0.01) and less steatosis on histology (p = 0.04). Consistently, animals from this group exhibited lower levels of serum aldosterone (p = 0.028) and lower hepatic triglyceride content (p = 0.008). This associated to a reduced expression of lipogenic genes, significant changes in lipid subspecies, lower HOMA-IR (p < 0.05), and lower expression of pro-inflammatory and profibrotic markers compared to those mice fed a Low-Na+/HFD. Additionally, mice fed a High-Na+/HFD showed higher expression of salt-inducible kinase (SIK)-1 and lower expression of serum-and-glucocorticoid-inducible kinase (SGK)-1. Similar results were observed with the MCD diet model.
ConclusionWe identified in two experimental models of NAFLD that High-Na+ diet content is associated to lower serum aldosterone levels and hepatic MR downregulation, associated to decreased steatosis and reduced de novo hepatic lipogenesis, proinflammatory and profibrotic markers. Decreased activation of hepatic MR seems to generate beneficial downstream inhibition of lipogenesis in experimental NAFLD.
Non-alcoholic fatty liver disease (NAFLD), the most common liver disease worldwide, is an heterogeneous disorder. The acronym groups different patient populations with a spectrum of liver histological abnormalities and different natural history and prognosis. The term non-alcoholic steatohepatitis (NASH) is applied when inflammation and evidence of hepatocellular injury (indicated by the presence of hepatocellular ballooning) are present. Of note, NASH is considered the more aggressive form of the disease that conveys a worse prognosis [1]. In the last decade, NAFLD became the focus of intense research due to the pandemic proportions it has reached (affecting almost a quarter of the world's population) [2,3]. Furthermore, NAFLD has been recognized as an independent risk factor for cardiovascular disease and type 2 diabetes mellitus (T2DM) as well as for some cancers [4–7].
Broadly, its pathogenesis is conceived as a sequential process where steatosis (pathological intracellular accumulation of lipids in the form of triglycerides) develops first, followed by local inflammation and triggering of a stereotyped fibrotic response to injury that contributes to progression to advanced liver fibrosis and cirrhosis [8]. Metabolic dysfunction reflected in the presence of metabolic syndrome (MetS) and insulin-resistance (IR) are considered central for NAFLD development and progression [2,9,10]. However, multiple pathophysiological pathways are involved and it is likely that they contribute differently in a given patient [11].
Dietary factors can contribute to steatosis development and eventually to disease progression [12]. Saturated fats and carbohydrates, particularly fructose, stimulate hepatic de novo lipogenesis, being associated with increased hepatic lipid content, inflammation, and possibly fibrosis [13]. Nutritional evaluation of patients with NAFLD had shown that they follow a western diet with a high content of fat and sodium and a suboptimal intake of micronutrients [14]. High sodium has been associated with increased glucocorticoids production, IR, pro-inflammatory cytokines and MetS in humans [15]. In obese mice, dietary sodium regulates the expression of adiponectin and sodium restriction reduces the profile of proinflammatory adipokines [16]. Another relevant protein regulated by sodium intake is the mineralocorticoid receptor (MR). The MR, normally activated by aldosterone and modulated by sodium intake, is expressed mainly in kidneys and heart [17]. MR activation is associated with fibrosis in target organs and also promotes inflammation, cardiovascular dysfunction, and adipose tissue differentiation [18]. Interestingly, MR blockade with specific antagonists like spironolactone or eplerenone ameliorated fibrogenesis in heart and kidney [19,20]. Liver MR is involved in hepatic fibrogenesis [21], which is potentially relevant for NAFLD progression. Previous work have shown that local activation of MR induces hepatocellular injury [22] and MR blockade with spironolactone or eplerenone decreased liver fibrosis in experimental models [21]. However, data on how sodium modulates hepatic MR expression is scarce [17,23].
Changes in sodium intake can modulate MR levels, pro-inflammatory cytokines and adiponectin in obese mice [16,17,23]. Also, chronic high sodium intake has been linked to MetS and NAFLD beyond the well-known effect in hypertension [15,24]. Shorter modifications of sodium with restricted intake can increase acutely aldosterone levels and be associated, counterintuitively, to metabolic dysregulation [25] and IR. Studying how sodium intake, MR activation and other factors that modulate liver damage is important because it can help identify potential therapeutic targets and long-term new approaches/treatments for clinical use [26]. In this work, we assess the effect over MR expression and NAFLD development by modulating sodium intake in two dietetic NAFLD mice models, focusing the activation of lipogenesis and lipid storage, inflammation and liver fibrosis. First, we will evaluate the effect in the HFD-fed mice model which is a more physiological model that mimics the feeding behavior observed in western populations. And second, we will perform our experimental approach using the MCD-fed mice which reproduce the most important hallmarks in NAFLD to NASH progression like hepatic inflammatory infiltration but lacking the extrahepatic features observed in most NAFLD individuals like obesity and insulin resistance.
2Material and methods2.1Animals and dietMale mice of 10 weeks (C57BL/6J, Jackson Laboratories, Bar Harbor, Maine) were used in accordance with protocols established by the ethical committee of our institution. Each animal was kept in an individual polycarbonate cage with controlled light and temperature. At the beginning of the study all animals were fed with a standard diet (Prolab 3000, Purina, PMI Feeds Inc., St. Louis MO). Two dietetics models were used to study NAFLD progression: High-fat diet (HFD) with 60% of calories provided by fat (#D12492, Research diets Inc., New Brunswick, NJ) for 12 weeks; and methionine-choline deficient (MCD) diet with 60 kcal% fat (# A06071302, Research diets Inc., New Brunswick, NJ) for 6 weeks. Sodium concentration was modified in each diet obtaining: (0.3%) (Normo-Na+), high sodium (3%) (High-Na+) and low sodium content (0.03%) (Low-Na+) [27]. Mice were grouped (n = 10) as follow: High-Na+/HFD, Normo-Na+/HFD, Low-Na+/HFD, High-Na+/MCD, Low-Na+/MCD. The results were compared to a control group consisting of mice fed with chow diet that with standard sodium content (0.3%). Mice that did not respond to HFD in terms of weight gaining were excluded from the study. Mice were euthanized through bleeding under generalized anesthesia. Samples from serum, liver, kidney, muscles and adipose tissue were taken and flash frozen to -80 °C until further analysis.
2.2Laboratory parametersAlanine aminotransferase levels (ALT) was measured using a Kovalent kit (Rio de Janeiro, Brazil) and results were expressed as IU/L. Serum triglyceride was measured through commercial kits (Sigma, St. Louis, MO). Adiponectin was measured through ELISA (Quantikine™ R&D Systems, Minneapolis, MN). Aldosterone was measured through Alpha Diagnostic International ELISA (San Antonio, Texas). Glycemia and insulin levels were measured with human kits (Wiesbaden, Germany). HOMA-IR (Homeostasis Model Assessment-Insulin resistance) was calculated with the formula fasting glycemia mg/dL × fasting insulin level ng/mL/22.5. Hepatic triglycerides were measured through the Folch method from 1.5 mL of homogenized liver tissue mixed with CHCl3-CH3OH (2:1, v/v).
2.3Histologic analysisLiver sections from the right lobe of all mouse livers were routinely fixed in 10% formalin and embedded in paraffin. Then 4 µm tissue sections were stained with hematoxylin/eosin and Sirius-red staining. Nuclei were counterstained with Hematoxylin. A blind investigator assigned a score for steatosis and inflammation as described [28]. Scores were given as it follows: Steatosis: grade 0, none present; grade 1, steatosis of <25% of parenchyma; grade 2, steatosis of 26–50% of parenchyma; grade 3, steatosis of 51–75% of parenchyma; grade 4, steatosis of >76% of parenchyma and inflammation: grade 0, no inflammatory foci; grade 1, 1–5 inflammatory foci per high power field; grade 2, >5 inflammatory foci/high power field. Liver fibrosis was quantified using digital image analysis of the red-stained area in Sirius red stained samples (ImageJ, NIH, USA).
2.4Real-time PCRTotal RNA from hepatic tissue was isolated using Speed Vacuum Total RNA Isolation System (Promega Corporation, Madison, WI, USA). Then it was quantified with a Nanodrop ND-1000 spectrophotometer. Reverse transcription (RT) was performed using 1 ug of total RNA, Quantitative Real Time PCR (qRT-PCR) was carried out in a StepOnePlus™ (96-well) PCR System (Applied Biosystems, Thermofisher, Waltham, MA, USA) using SYBR Green method. The sequences of the primer sets are described in the Supplementary materials file. mRNA levels were normalized to the housekeeping gene 18S and further normalized to the mean expression level of the control group. Relative gene expression was calculated via the 2 ddct. The genes studied are described in Suppl. Table 1.
2.5Lipid contentFor analysis of liver lipid concentration, lipids were extracted by the method described by Folch et al. [29] and then, they were separated by thin layer chromatography as described by Ruiz & Ochoa [30]. Free fatty acid and triglyceride levels were determined using commercial Kits (Menarini Diagnostics, Italy) from hepatic homogenate and lipid extract, respectively. Liver concentration of diglycerides, different species of cholesterol and glycerophospholipids were determined using a GS-800 densitometer and Quantity One software (Bio Rad, USA).
2.6Statistical analysisAll results are expressed as mean with standard deviation. Two tailed non-paired student t-test was used to compare results between groups. Mann-Whitney U test or Kruskal-Wallis test was used to compare 2 or more groups. Significant p-value ≤0.05. ANOVA was used for comparison parametric variables between multiple groups. Post-hoc analysis using Bonferroni or Dunn was used to correct for multiple comparisons. Histologic variables were expressed as percentages and Chi-Square test was used to compare groups. All data were analyzed and graphed with GraphPad 8 Prism (GraphPad Software, Inc).
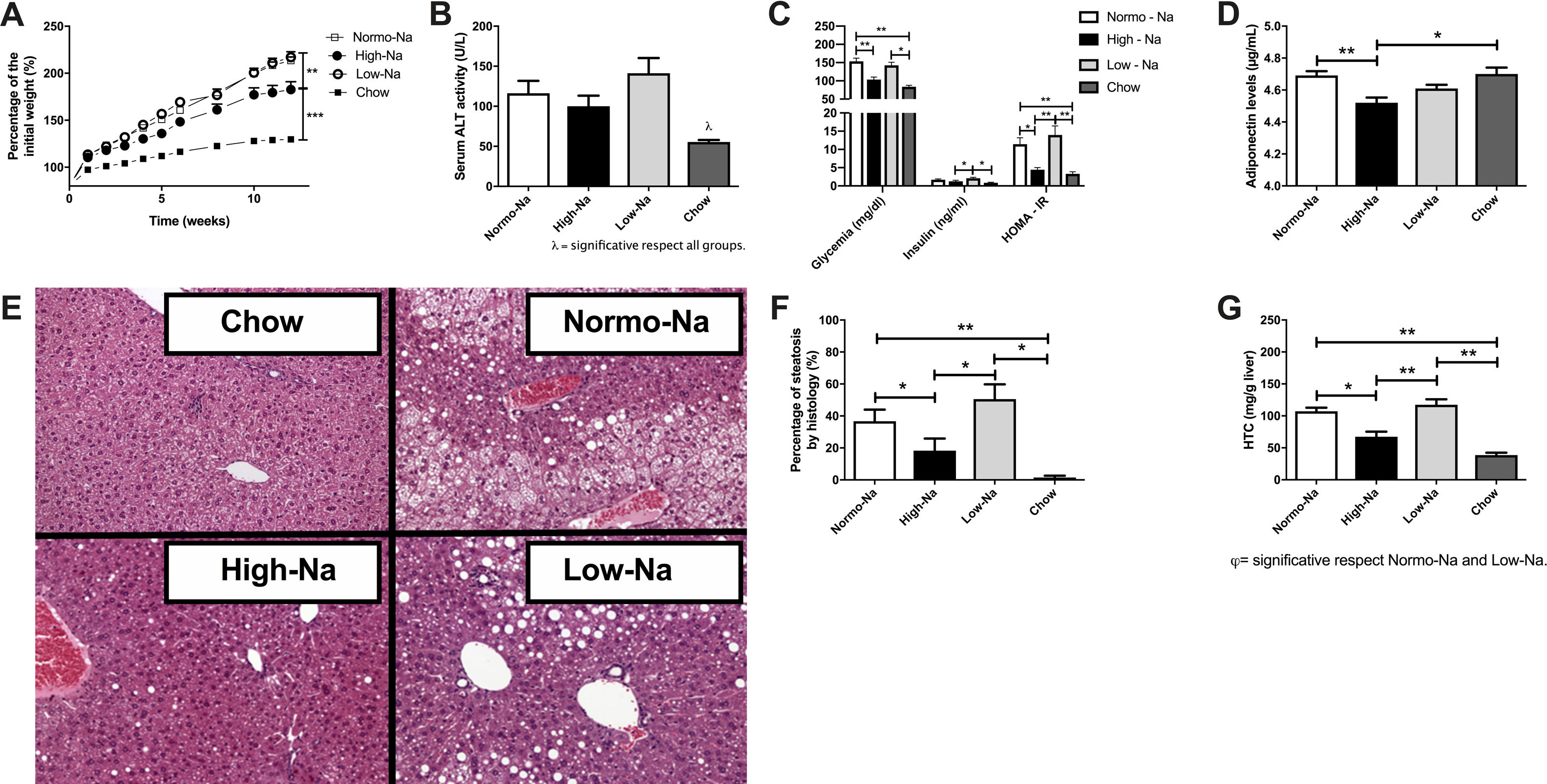
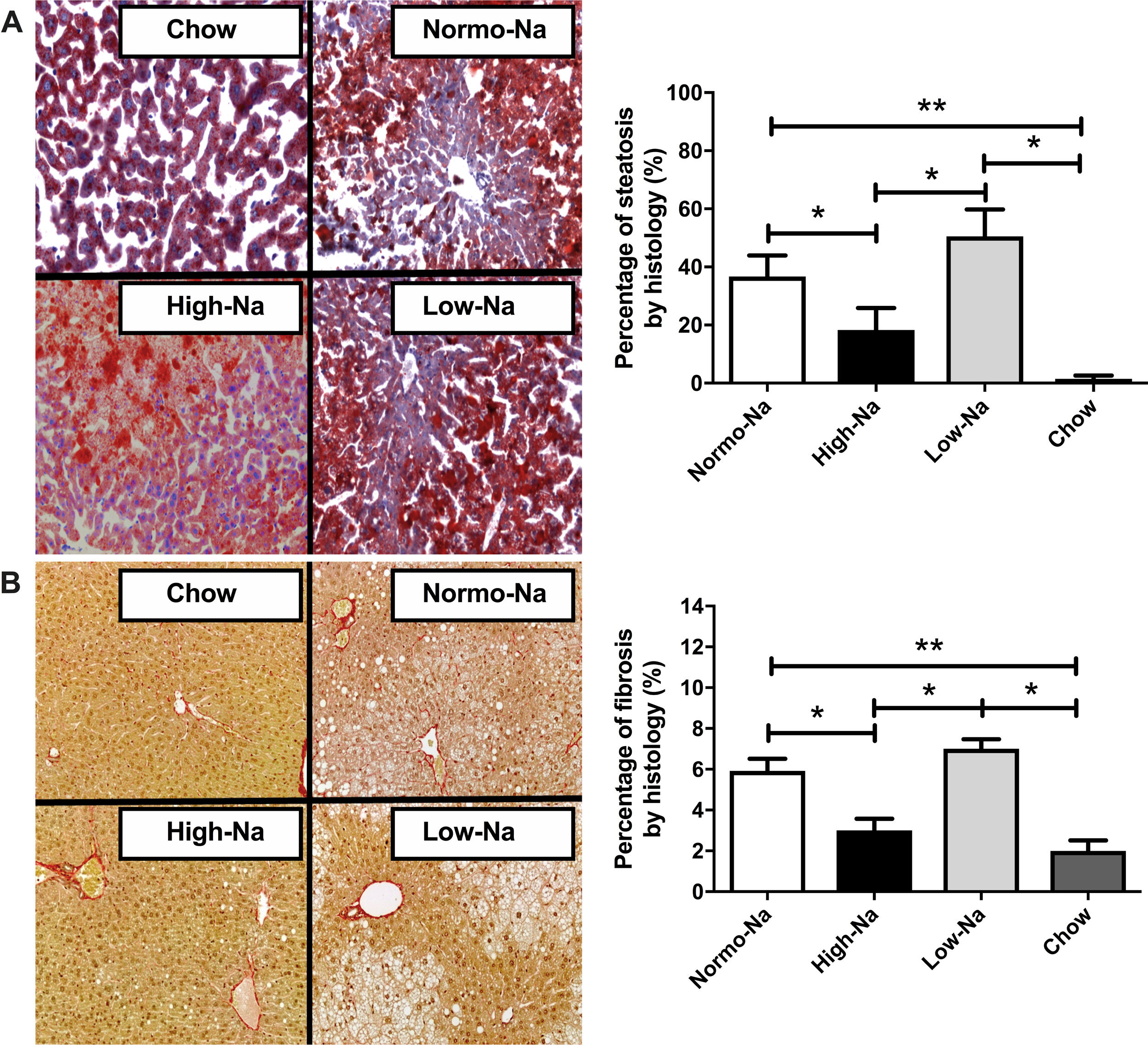
3Results3.1Effect of HFD with different sodium content on liver steatosis and IRWe used a HFD mice model of NAFLD, which in spite of being a mild-intensity model most closely mimics the pathophysiology of human NAFLD with IR and MetS [31]. Fig. 1 shows the anthropometric and serum parameters after 12 week of experimental diet feeding. HFD-fed mice had a larger increase in body weight (p < 0,001), the High-Na+/HFD group had a slightly lower increase in body weight by the end of the experiment (Fig. 1A), despite consuming the same amount of food. The plasma levels of ALT were similar between the groups, only significant differences were found between the chow-fed group (55.5 ± 2.4 IU/L) and the Low-Na+/HFD group (141.1 ± 19 IU/L), (p = 0.01) (Fig. 1B). Fasting glycemia was higher in the groups with Normo-Na+/HFD (153.5 ± 8.6 mg/dL) and Low-Na+/HFD (142.5 ± 8.5 mg/dL), compared to the High-Na+/HFD group (103.3 ± 6.9 mg/dL, p = 0.008 with respect to Normo-Na+/HFD) and chow (83.75 ± 3.9 mg/dL) (Fig. 1C). Insulin levels showed a similar pattern, with a significant increase in the Low-Na+/HFD group (2.1 ± 0.26 ng/mL) compared to the High-Na+/HFD group (1.2 ± 0.3 ng/mL, p = 0.04) and chow (0.87 ± 0.12 ng/mL, p = 0.02) (Fig. 1C). Consistently, HOMA-IR was significantly higher in the Normo-Na+/HFD (11.4 ± 1.7) and Low-Na+/HFD (13.95 ± 2.5) groups compared to the High-Na+/HFD (4.44 ± 0.59, p < 0.05) and chow (3.2 ± 0.57, p < 0.05) groups (Fig. 1C). On the other hand, plasma adiponectin levels, an adipokine that upregulates lipid oxidation and catabolism, were lower in the High-Na+/HFD group (4.52 ± 0.03 µg/mL) compared to mice fed Normo-Na+/HFD (4.69 ± 0.02 µg/mL, p = 0.003) and chow (4.7 ± 0.04 µg/mL, p = 0.03) (Fig. 1D). To assess the impact of our intervention on liver histology, we assessed liver tissue for steatosis and fibrosis. Evaluation of the liver parenchyma with H&E staining showed a higher percentage of liver steatosis in the Low-Na+/HFD group (50.5 ± 9.2%) compared to the High-Na+/HFD group (18.3 ± 7.59%, p = 0.04) and chow (0%, p = 0.01) (Fig. 1E and F). Evaluation of hepatic triglyceride content confirmed histology findings. Mice fed High-Na+/HFD had a lower hepatic triglyceride content (67.38 ± 7.85 mg/g of liver) compared to Normo-Na+/HFD (107.3 ± 5.56 mg/g of liver), p = 0.04) and Low-Na+/HFD groups (117.4 ± 8.57 mg/g of liver, p = 0.008). In addition, as expected, the lowest amount of liver triglycerides (38.73 ± 3.75 mg/g of liver) was observed in the chow group, not being different from the High-Na+/HFD group (Fig. 1G). This finding was confirmed by evaluating histological sections by staining Oil-red-O, where the High-Na+/HFD group showed significantly lower liver steatosis (24.9%) than Low-Na+/HFD (62.4%) or Normo-Na+/HFD (37.7%) (Fig. 2A). No differences in fibrosis were observed by Sirius-red staining (5.5% in the High-Na+/HFD vs. 5.8% in the Low-Na+/HFD group) (Fig. 2B). In summary, we demonstrated that mice fed with High-Na+/HFD developed less steatosis, MetS and IR, compared to Normo-Na+/HFD and Low-Na+/HFD groups.
 with different Na+ content on anthropometric parameters and metabolic markers on mice. A) High-Na+/HFD induced less weight gain compared to Normo-Na+/HFD and Low-Na+/HFD. B) There were no significant differences in plasma alanine aminotransferase (ALT) levels between HFD with different Na+ content. C) High-Na+/HFD induced less fasting glycemia, insulin and HOMA-IR than Low-Na+/HFD. D) High-Na+/HFD induced less plasma adiponectin levels than Normo-Na+/HFD. E) Representative images from histologic samples stained with H&E showing that High-Na+/HFD induced less hepatic steatosis than Low-Na+/HFD. F) Percentage of steatosis per field. G) High-Na+/HFD induced less hepatic triglyceride expression than Normo-Na+/HFD and Low-Na+/HFD. n = 10 mice per group, *p < 0.05, ** p < 0.01 by ANOVA.")
Effect of High fat diet (HFD) with different Na+ content on anthropometric parameters and metabolic markers on mice. A) High-Na+/HFD induced less weight gain compared to Normo-Na+/HFD and Low-Na+/HFD. B) There were no significant differences in plasma alanine aminotransferase (ALT) levels between HFD with different Na+ content. C) High-Na+/HFD induced less fasting glycemia, insulin and HOMA-IR than Low-Na+/HFD. D) High-Na+/HFD induced less plasma adiponectin levels than Normo-Na+/HFD. E) Representative images from histologic samples stained with H&E showing that High-Na+/HFD induced less hepatic steatosis than Low-Na+/HFD. F) Percentage of steatosis per field. G) High-Na+/HFD induced less hepatic triglyceride expression than Normo-Na+/HFD and Low-Na+/HFD. n = 10 mice per group, *p < 0.05, ** p < 0.01 by ANOVA.
 High-Na+/HFD induced less percentage of steatosis than Low-Na+/HFD measured using Oil-Red-O Staining 40×. B) There were no significant differences in percentage of fibrosis between HFD with different Na+ content using Sirius Red staining 40×. n = 10 mice per group, *p < 0.05, by ANOVA.")
Effect of HFD with different Na+ content on liver histology. Representative pictures of Hematoxylin and Eosin, Oil-red and Sirius red staining of liver sections. A) High-Na+/HFD induced less percentage of steatosis than Low-Na+/HFD measured using Oil-Red-O Staining 40×. B) There were no significant differences in percentage of fibrosis between HFD with different Na+ content using Sirius Red staining 40×. n = 10 mice per group, *p < 0.05, by ANOVA.
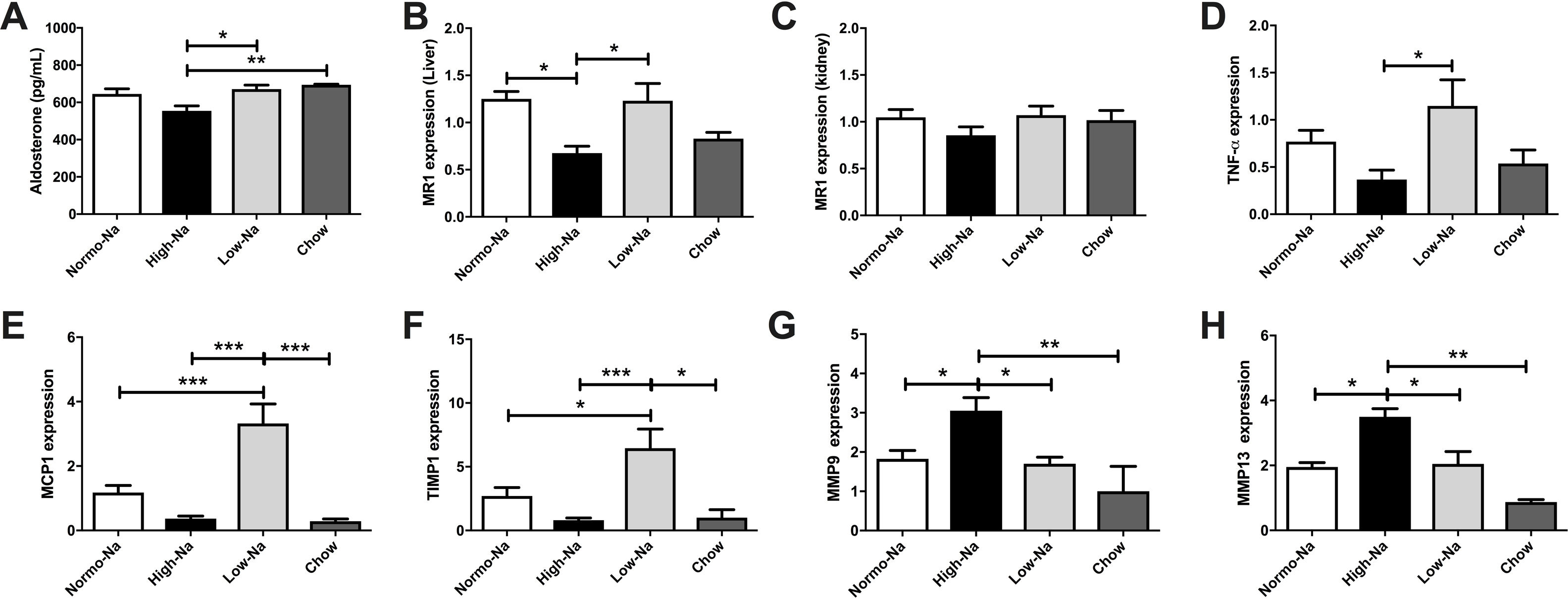
We hypothesize that secondary to chronic High-Na + exposure reduces aldosterone availability and hepatic MR is downregulated.Serum aldosterone levels were lower in High-Na+/HFD-fed mice (554.3 ± 27.28 pg/mL) compared to Low-Na+/HFD-fed mice (671.4 ± 21.93 pg/mL, p = 0.028) and chow (695 ± 2.69 pg/mL, p = 0.007) (Fig. 3A). This suggests that in our model aldosterone is mainly regulated by sodium intake, independent of lipid ingestion. Assessment of mRNA expression of MR in liver samples, showed that the group fed with High-Na+/HFD had a lower relative expression with respect to the groups fed with Normo-Na+/HFD (p ≤ 0.01) and Low-Na+/HFD (p ≤ 0.01) (Fig. 3B). In contrast, the expression of MR in the kidney showed no differences between the experimental groups (p = 0.32) (Fig. 3C). This suggests that MR expression in the kidney is regulated differently and that there is a local modulation of serum aldosterone levels and MR expression in the liver.
 High-Na+/HFD showed less serum aldosterone level than Low-Na+/HFD. B) High-Na+/HFD induced less hepatic MR expression than Normo-Na+/HFD and Low-Na+/HFD. C) There was no significant difference in kidney MR expression between HFD with different Na+ content. D) High-Na+/HFD induced less serum TNF-a expression than Low-Na+/HFD. E) High-Na+/HFD induced less serum MCP-1 expression than Low-Na+/HFD. F) High-Na+/HFD induced less serum TIMP-1 expression than Low-Na+/HFD. G) High-Na+/HFD induced MMP9 expression than Low-Na+/HFD. H) High-Na+/HFD induced MMP13 expression than Low-Na+/HFD n = 10 mice per group, *p < 0.05, ** p < 0.01, ***p < 0.001.")
Down-modulation of the mineralocorticoid pathway by High-Na+/HFD in the liver. A) High-Na+/HFD showed less serum aldosterone level than Low-Na+/HFD. B) High-Na+/HFD induced less hepatic MR expression than Normo-Na+/HFD and Low-Na+/HFD. C) There was no significant difference in kidney MR expression between HFD with different Na+ content. D) High-Na+/HFD induced less serum TNF-a expression than Low-Na+/HFD. E) High-Na+/HFD induced less serum MCP-1 expression than Low-Na+/HFD. F) High-Na+/HFD induced less serum TIMP-1 expression than Low-Na+/HFD. G) High-Na+/HFD induced MMP9 expression than Low-Na+/HFD. H) High-Na+/HFD induced MMP13 expression than Low-Na+/HFD n = 10 mice per group, *p < 0.05, ** p < 0.01, ***p < 0.001.
Next, we assessed hepatic expression of selected inflammation and fibrosis-related genes to better evaluate the impact of the dietary sodium changes in the HFD model of steatohepatitis. Low-Na+/HFD -fed mice exhibited a higher TNF-α mRNA hepatic mRNA levels were significantly higher than the other experimental groups (Fig. 3E). Regarding fibrosis, we tested the levels of TIMP-1, which were decreased in mice fed with high sodium (Fig. 3F). While the levels of MMP9 and MMP13 were found augmented (Fig. 3G and H). These findings, suggest that, in the presence of steatosis, changes in sodium intake modulates inflammation and fibrosis.
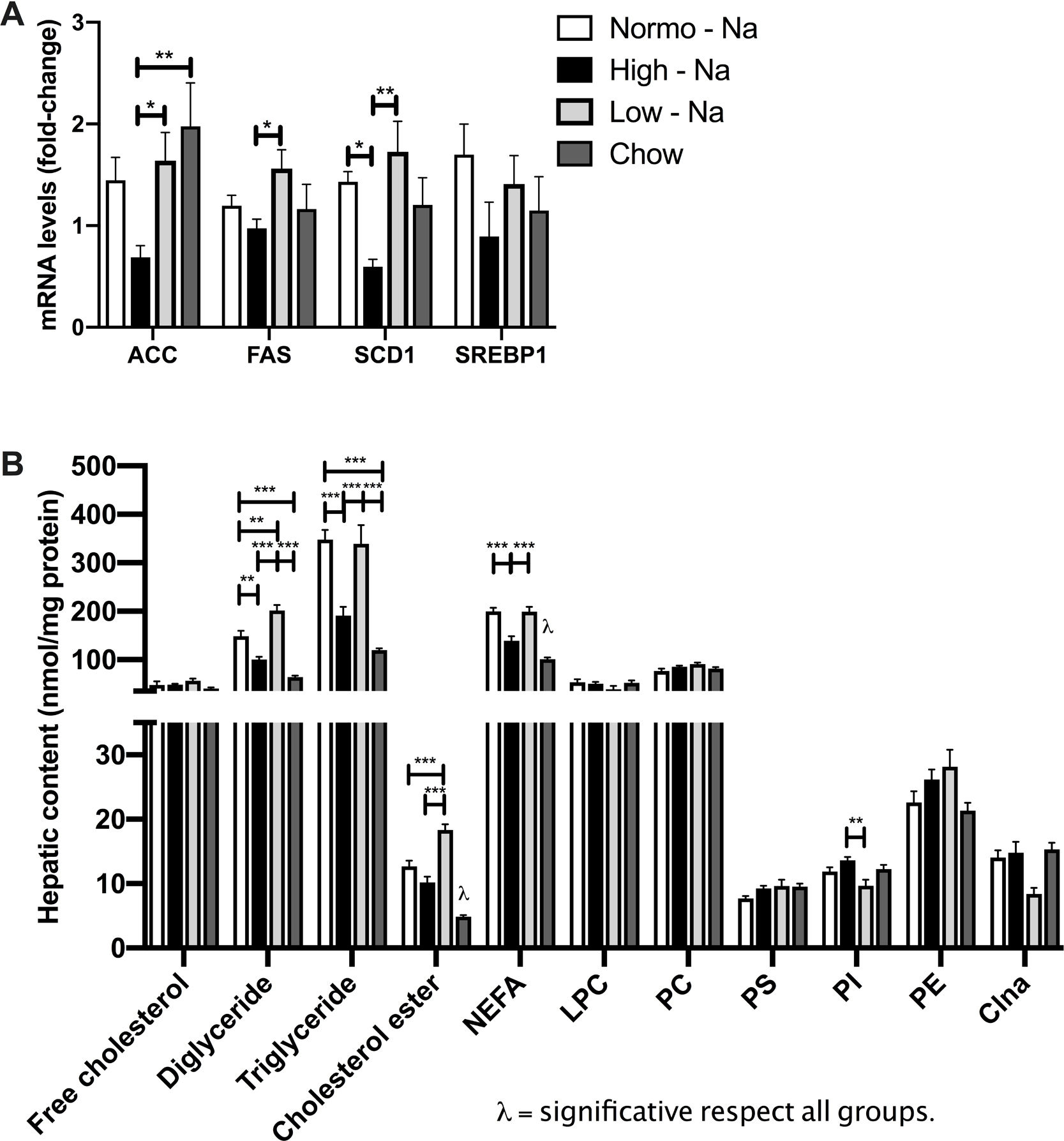
3.4Effect of HFD with different content of sodium in liver lipids sub-species and lipogenesisIn order to assess if sodium intake modulates liver lipid species accumulating during steatosis, we extracted liver lipids by chromatography and quantified them with optic densitometry. We observed significant differences in the liver content of neutral lipids with diglycerides being higher in Low-Na+/HFD-fed mice (201.6 ± 11.07 nmol/mg protein) compared to Normo-Na+/HFD-fed mice (148.5 ± 11.37 nmol/mg protein, p = 0.003) and High-Na+/HFD-fed (100.6 ± 5.36 nmol/mg protein, p < 0.0001) and chow (63.75 ± 3.64 nmol/mg protein, p < 0.0001) (Fig. 4A). Hepatic triglycerides content was higher in the mice under Low-Na+/HFD-fed (372.4 ± 20.33 nmol/mg protein) and Normo-Na+/HFD-fed (347.7 ± 20.11 nmol/mg protein), compared to High-Na+/HFD-fed mice (190.6 ± 18.42 nmol/mg protein, p < 0.0001) and chow (119.8 ± 3.4 nmol/mg protein, p < 0.0001)) (Fig. 4A). Total liver non-esterified fatty acid concentration (NEFA) was lower in the group of mice fed High-Na+/HFD (139.4 ± 8.9 nmol/mg protein), compared to mice fed Normo-Na+/HFD (199.9 ± 7.1 nmol/mg protein, p = 0.0003) and Low-Na+/HFD (199.1 ± 9.9 nmol/mg protein, p = 0.0004) and higher than the chow group (100.8 ± 3.7 nmol/mg protein, p = 0.045)) (Fig. 4A). Regarding sterols, not changes were observed in free cholesterol, but hepatic cholesterol ester content was higher in mice under Low-Na+/HFD (18.32 ± 0.89 nmol/mg protein), compared to mice fed Normo-Na+/HFD (12.68 ± 0.88 nmol/mg protein, p = 0.0009), High-Na+/HFD (10.17 ± 0.93 nmol/mg protein, p < 0.0001) and chow (4.85 ± 0.25 nmol/mg protein, p < 0.0001) (Fig. 4B). With regard to glycerophospholipids (phosphatidylcholine, phosphatidylethanolamine, lysophosphatidylcholine, phosphatidylserine, phosphatidylinositol, and cardiolipin) no differences were observed (Fig. 4C) across the experimental groups. Only, phosphatidylinositol showed a higher content in mice fed High-Na+/HFD (13.63 ± 0.51 nmol/mg protein), compared to mice fed Low-Na+/HFD (9.68 ± 0.9 nmol/mg protein, p = 0.003) (Fig. 4C).
 High-Na+/HFD induces less content of neutral lipids (triglycerides, diglycerides and fatty acids) determined by chromatography. B) High-Na+/HFD induces significantly less content of cholesterol ester but no difference in free cholesterol. C) High-Na+/HFD does not induce less content of glycerophospholipids (phosphatidylcholine, phosphatidylethanolamine, lysophosphatidylcholine, phosphatidylserine, phosphatidylinositol, cardiolipin). D) High-Na+/HFD induced less expression of most lipogenic enzymes, with the exception of SREBP1. n = 10 mice per group, *p < 0.05, ** p < 0.01, ***p < 0.001 by ANOVA.")
Effect of HFD with different Na+ content of lipid species. A) High-Na+/HFD induces less content of neutral lipids (triglycerides, diglycerides and fatty acids) determined by chromatography. B) High-Na+/HFD induces significantly less content of cholesterol ester but no difference in free cholesterol. C) High-Na+/HFD does not induce less content of glycerophospholipids (phosphatidylcholine, phosphatidylethanolamine, lysophosphatidylcholine, phosphatidylserine, phosphatidylinositol, cardiolipin). D) High-Na+/HFD induced less expression of most lipogenic enzymes, with the exception of SREBP1. n = 10 mice per group, *p < 0.05, ** p < 0.01, ***p < 0.001 by ANOVA.
Assessment of the mRNA expression of key hepatic lipogenic enzymes was carried out in samples from different experimental groups in order to explore if dietary sodium intake modulates lipid biosynthesis in liver tissue. Expression of Acetyl-CoA carboxylase (ACC), Fatty acid synthase enzyme (FAS) and Stearoyl-CoA desaturase 1 (SCD1) was lower in mice fed High-Na+/HFD (0.68 ± 0.11) compared to mice fed Low-Na+/HFD (Fig. 4D) suggesting that in the context of steatosis, dietary sodium modulates lipogenesis in agreement with previous observations [32]. No differences were observed when evaluating mRNA levels Sterol regulatory element-binding protein 1 (SREBP1) (p = 0.68) (Fig. 4D).
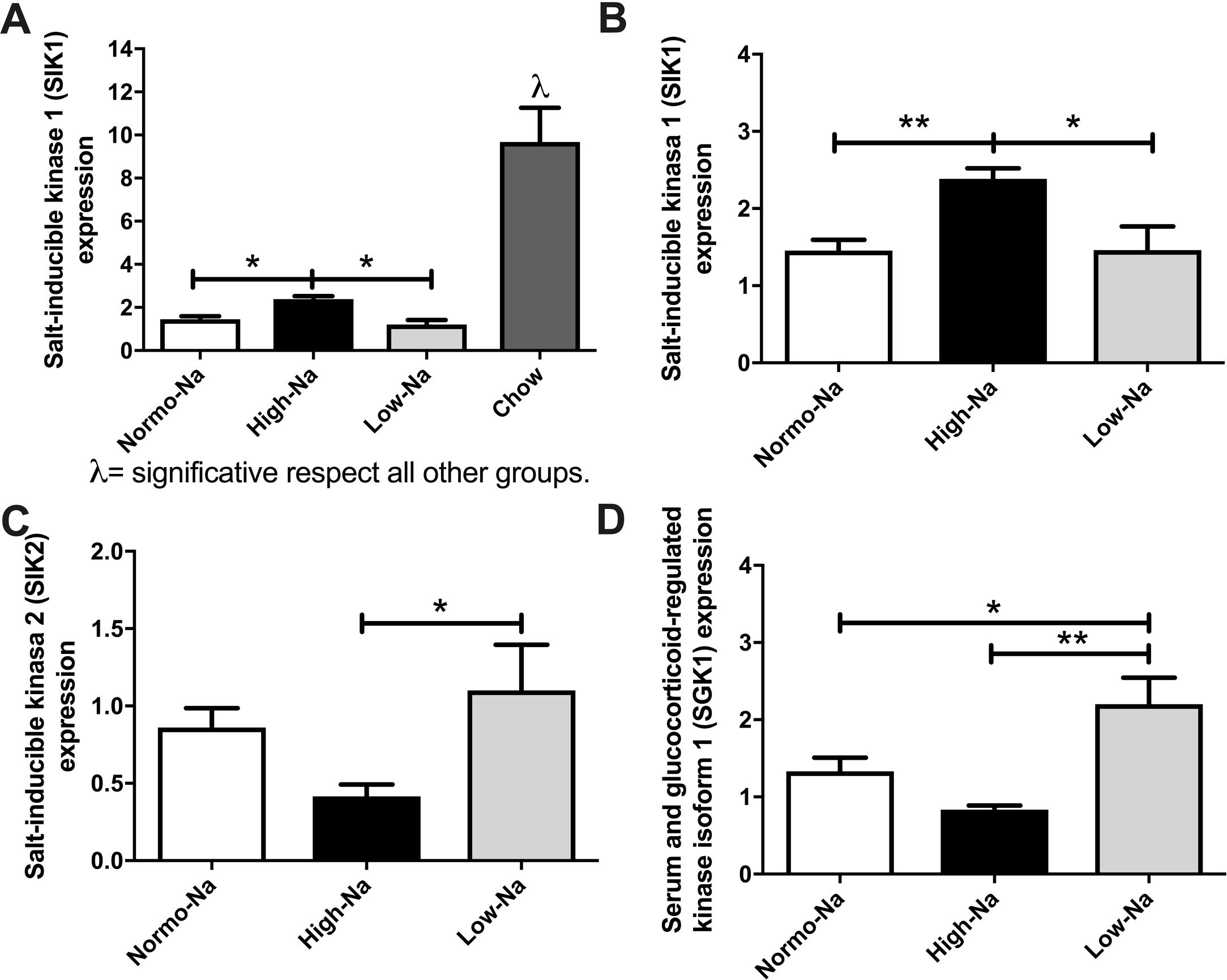
3.5Effect of HFD with different sodium content of on salt-inducible kinases (SIK) and serum-and-glucocorticoid-inducible kinase 1 (SGK1) enzymesAiming to gain insights on the mechanisms by which the sodium/aldosterone/MR axis could influence hepatic lipogenesis, we next explored if sodium content of HFD modulates the hepatic expression of salt-inducible kinases (SIKs). In our model, SIK1 expression was significantly lower in’ groups of mice fed with HFD compared to chow (Fig. 5A). Of note, SIK1 expression was significantly higher in mice fed with High-Na+/HFD compared to Low-Na+/HFD and Normo-Na+/HFD (Fig. 5B). This suggests that SIK1 expression is regulated by lipid content and sodium content of diet. The expression of SIK2 was significantly lower in High-Na+/HFD compared to Low-Na+/HFD (Fig. 5C). Finally, we assessed the hepatic expression of the enzyme serum-and-glucocorticoid-inducible kinase 1 (SGK1), observing a downregulation of SGK1 with High-Na+/HFD and an upregulation with Low-Na+/HFD (Fig. 5D).
 High-Na+/HFD induced more SIK-1 expression than low and Normo-Na+/HFD. B) High-Na+/HFD induced more SIK-1 expression than Low-Na+/HFD and Normo-Na+/HFD. C) High-Na+/HFD induced less SIK-2 expression than Low-Na+/HFD. D) High-Na+/HFD induced less SGK1 expression than Low-Na+/HFD. n = 10 mice per group, *p < 0.05, **p < 0.01 by ANOVA.")
Effect of HFD and MCD with different concentration of Na+ on expression of salt inducible kinases and illustration of the proposed pathophysiological mechanism of how MR activation modifies hepatic lipogenesis. A) High-Na+/HFD induced more SIK-1 expression than low and Normo-Na+/HFD. B) High-Na+/HFD induced more SIK-1 expression than Low-Na+/HFD and Normo-Na+/HFD. C) High-Na+/HFD induced less SIK-2 expression than Low-Na+/HFD. D) High-Na+/HFD induced less SGK1 expression than Low-Na+/HFD. n = 10 mice per group, *p < 0.05, **p < 0.01 by ANOVA.
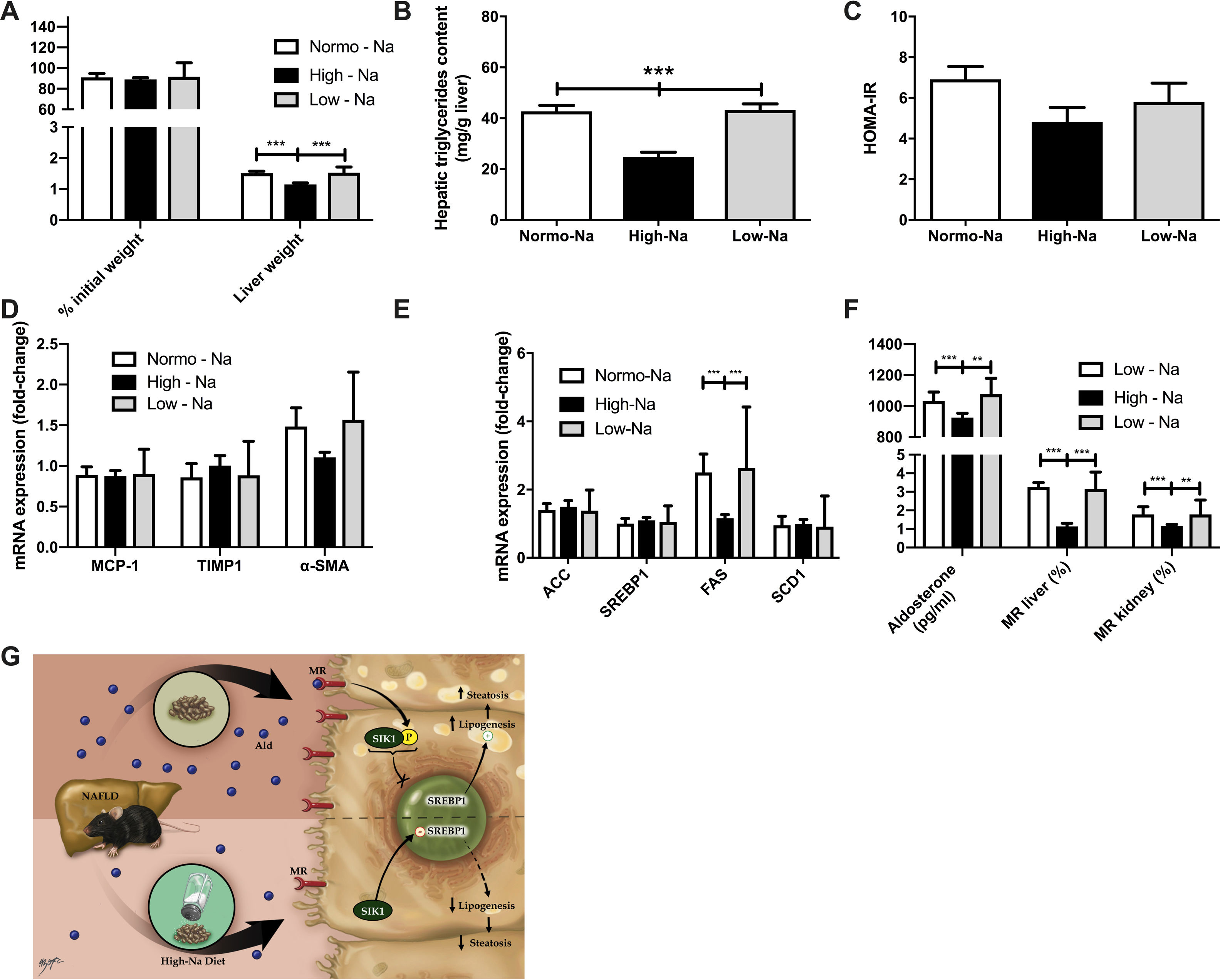
Finally, we used the MCD diet for 6 weeks as a second NAFLD/NASH mouse model to explore the effects of modulation of MR by dietary sodium on liver damage; this model of NASH is not associated with significant IR but associated with inflammation and fibrosis. We used Mice fed MCD diet did not show significant weight differences, expressed as a percentage of initial weight, at the end of 6 weeks between the High-Na+/MCD (88.92 ± 1.77%) and Low-Na+/MCD (91.55 ± 4.0%, p = 0.79) (Fig. 6A). On the other hand, liver mass was lower in the group fed High-Na+/MCD (1.14 ± 0.04 g) compared to the group fed Low-Na+/MCD (1.52 ± 0.05 g, p < 0.0001) (Fig. 6A). Of note, hepatic triglyceride content in mice fed High-Na+/MCD was significantly lower than that of mice fed a Low-Na+/MCD ((24.83 ± 1.8 mg/g liver vs. 43.25 ± 2.3 mg/g of liver, p < 0.0001) (Fig. 6B) similar to that we observed in animals fed HFD with different sodium contents. In the MCD fed-mice. Additionally, HOMA was similar between these two groups (p = 0.37) (Fig. 6C). To assess inflammation, MCP-1 tissue expression was evaluated showing no statistically significant difference between the group of mice fed High-Na+/MCD (0.87 ± 0.06) and those fed Low-Na+/MCD (0.90 ± 0.1, p = 0.86) (Fig. 6D). Unlike what was observed with the HFD diet, fibrosis markers TIMP1 and α-SMA expression did not show differences between groups (Fig. 6D). Regarding lipogenesis, it was observed that the expression of Fatty acid synthase (FAS) was lower in mice fed High-Na+/MCD (1.1 ± 0.3) compared to mice fed Low-Na+/MCD (2.6 ± 0.36, p = 0.0005), without evidencing statistical difference in the other enzymes involved in lipogenesis (Fig. 6E). As expected, plasma aldosterone levels were lower in the group of mice fed High-Na+/MCD (925.3 ± 28.5 pg/mL) compared to mice fed Low-Na+/MCD (1076 ± 36.36 pg/mL, p = 0.011) (Fig. 6F). Additionally, MR expression in both liver and kidney was significantly lower in mice fed High-Na+/MCD compared to Low-Na+/MCD-fed mice (Fig. 6F).
 There was no significant difference in increase of weight in mice fed with MCD, however, there is significantly less liver weight gain in the High-Na+/MCD. B) High-Na+/MCD showed less percentage of hepatic triglycerides content than Low-Na+/MCD. C) There were not significant differences in HOMA-IR calculated between the different MCD groups. D) There were no significant differences between different MCD groups in expression of fibrosis markers including monocyte chemoattractant protein-1, tissue inhibitor of metalloproteinases-1, alpha smooth muscle actin. E) There were not significant differences between different MCD groups in lipid genes activation such as ACC, SREBP1, SCD1. There is significantly less expression of FAS in High-Na+/MCD compared to Low-Na+/MCD. F) High-Na+/MCD activates mineralocorticoid receptor signaling significantly less than Low-Na+/MCD, with less serum aldosterone level, hepatic mineralocorticoid receptor (MR) activation and kidney MR activation. n = 10 mice per group, *p < 0.05, ** p < 0.01, ***p < 0.001 by ANOVA. G) Summary of the proposed mechanism. We propose that a high-Na+ intake leads to lower levels of MR expression, lower levels of aldosterone and less activation of RAAS. This down-modulation of the mineralocorticoid pathway allows for SIK1 overexpression. SIK-1 translocate to the nucleus where it downregulates the expression of lipogenic enzymes -such as FAS, ACC, SCD1-downstream of SREBP1, resulting in less lipogenesis, therefore less steatosis. Meanwhile, in the livers of animals with normal or low-Na+ intake, a HFD induces higher levels of aldosterone, activating the mineralocorticoid pathway, therefore, causing SIK-1 phosphorylation, phosphorylated SIK-1 accumulates in the cytoplasm and doesn’t take part in the regulation of hepatic lipogenic gene expression.")
Effect of MCD with high and low Na+ concentration on hepatic steatosis and activation of mineralocorticoid receptor signaling. A) There was no significant difference in increase of weight in mice fed with MCD, however, there is significantly less liver weight gain in the High-Na+/MCD. B) High-Na+/MCD showed less percentage of hepatic triglycerides content than Low-Na+/MCD. C) There were not significant differences in HOMA-IR calculated between the different MCD groups. D) There were no significant differences between different MCD groups in expression of fibrosis markers including monocyte chemoattractant protein-1, tissue inhibitor of metalloproteinases-1, alpha smooth muscle actin. E) There were not significant differences between different MCD groups in lipid genes activation such as ACC, SREBP1, SCD1. There is significantly less expression of FAS in High-Na+/MCD compared to Low-Na+/MCD. F) High-Na+/MCD activates mineralocorticoid receptor signaling significantly less than Low-Na+/MCD, with less serum aldosterone level, hepatic mineralocorticoid receptor (MR) activation and kidney MR activation. n = 10 mice per group, *p < 0.05, ** p < 0.01, ***p < 0.001 by ANOVA. G) Summary of the proposed mechanism. We propose that a high-Na+ intake leads to lower levels of MR expression, lower levels of aldosterone and less activation of RAAS. This down-modulation of the mineralocorticoid pathway allows for SIK1 overexpression. SIK-1 translocate to the nucleus where it downregulates the expression of lipogenic enzymes -such as FAS, ACC, SCD1-downstream of SREBP1, resulting in less lipogenesis, therefore less steatosis. Meanwhile, in the livers of animals with normal or low-Na+ intake, a HFD induces higher levels of aldosterone, activating the mineralocorticoid pathway, therefore, causing SIK-1 phosphorylation, phosphorylated SIK-1 accumulates in the cytoplasm and doesn’t take part in the regulation of hepatic lipogenic gene expression.
We studied the effect of MR modulation by sodium intake on liver injury in 2 different experimental models of NAFLD and made several novel observations: 1) mice fed with High-Na+/HFD developed less steatosis, MetS and IR than the comparison groups; 2) 12 week administration of a High-Na+/HFD consistently decreased hepatic aldosterone and MR expression levels 3) Lower MR expression was associated with less steatosis, inflammation and fibrosis markers; 4) variation in sodium content of HFD was associated with significant changes in hepatic lipidic profile and modulated the expression of enzymes involved in hepatic lipogenesis; and 5) SIK1 and SGK1 were modulated by sodium content of HFD, which could be related to changes in hepatic lipogenesis. Most of the changes observed in the classic HFD-induced NAFLD mouse model were recapitulated in mice with MCD-induced NASH. Collectively, our data suggest that sodium/aldosterone/MR pathway could play a role in modulating NAFLD development and progression.
We identified that hepatic MR modulation, rather than the dietary sodium content by itself, may regulate the development and severity of NAFLD in mice. We observed that High-Na+/HFD-fed mice expressed lower levels of aldosterone (implying less RAAS activation) and subsequently lower hepatic MR levels, they also displayed lower levels of glycemia, insulinemia and, therefore, HOMA-IR, compared to Low-Na+/HFD-fed mice. Interestingly, MR kidney expression was not affected, which could be explained by a renal adaptive response to chronic exposures (12-week diet). The lower expression of hepatic MR is a critical finding, since we recently described that an increased expression of MR in a murine NAFLD model is associated with increased inflammation and fibrosis and MR blockade with eplerenone has anti-steatotic and anti-fibrotic liver effects [21].Therefore, the High-Na+/HFD would have a similar effect than MR antagonists and this could explain the reduced steatosis observed in these models. In Low-Na+/HFD-fed mice, there was an induction of a state of secondary hyperaldosteronism and, therefore, an increase in the hepatic MR expression with the pro-steatotic, pro-inflammatory and pro-fibrotic effects. Hepatic MR activation is associated with more inflammation and fibrosis because RAAS activation has shown to cause liver damage through vasoactive and profibrotic proliferation [33]. Furthermore, MR blockage has shown to decrease adipose inflammation, decrease hepatic collagen deposition, and improve glucose metabolism [19].
These hypotheses are consistent with histological findings, where the High-Na+/HFD prevented steatosis, however, without evidencing significant differences in fibrosis based on the Sirius Red stain finding. This could be explained because the experimental model of NAFLD with HFD diet does not induce significant fibrosis [34]. On the other hand, when analyzing gene expression, it is evident that the group of mice fed with a High-Na+/HFD compared to the group of mice fed with a Low-Na+/HFD have lower levels of inflammation (TNF-α and MCP1) and fibrosis markers (TIMP1).
To better characterize the observed changes in steatosis, we set out to study liver lipogenesis pathways. We observed a lower expression of the main enzymes involved in lipogenesis (ACC, SCD1 and FAS) in High-Na+/HFD fed mice compared to the Low-Na+/HFD group. We did not observe differences in SREBP1, which could be determined by the short half-life of the mRNA of this transcription factor, however, its downstream enzymes (FAS, SCD1) were consistently modified [35].
Additionally, we evaluated lipids subtypes, observing a decreased triglyceride content in the group fed with High-Na+/HFD, this group also showed lower content of cholesterol esters and higher content of phosphatidylinositol compared to mice fed a Low-Na+/HFD. Mice fed a High-Na+/HFD had a lower content of diglycerides. This finding is key since diglycerides are associated with hepatic IR [36]. It should be noted that mice under High-Na+/HFD had a comparatively lower percentage of weight gain than mice in the Normo-Na+/HFD and Low-Na+/HFD groups, despite ingesting similar amounts of food. This has been previously described as a consequence of the effect of salt content on digestive efficiency [37]. High-Na + intake reduces the total food intake and appetite, therefore decreasing total body fat and the development of MetS and NAFLD [38]. Moreover, high-Na + diets have shown to decrease the storage of lipids within adipocytes by inhibiting adipocyte differentiation through upregulation of calcium pathways [38]. It is also interesting that the group of mice with a High-Na+/HFD shows slightly lower levels of adiponectin [16].
The MCD diet model of NAFLD constitutes a lean model of liver injury in which IR and MetS are not present [39] (41), unlike the HFD model. Consistent with the pathogenesis of the model, there were no significant differences in HOMA-IR or body weight. However, differences were observed in the liver mass, being lower in the High-Na+/MCD-fed mice, Similar to the HFD diet model, MCD showed lower levels of aldosterone in High-Na+/MCD-fed mice. In addition, lower hepatic triglyceride content and decrease in FAS were observed. No differences were observed in the pro-inflammatory and pro-fibrotic genes evaluated, probably due to the early nature of the experimental model (6 weeks).
We investigated downstream signaling, identifying the family of salt-inducible kinases (SIK1, SIK2 and SGK1) 26 that play a regulatory role in steroidogenesis [40,41], modulate inflammation, participate in cellular processes such as apoptosis and proliferation and have a role in multiple organs like adipose tissue, heart and kidney. In particular, SIK1 has shown to modulate lipogenesis through regulation of SREBP-1 [32]. Overexpression of SIK1 inhibits liver lipogenic genes expression and SIK-1 knockout mice greatly increases lipogenic genes expression [32], we observed that SIK1 expression was significantly higher in mice fed with High-Na+/HFD compared to Low-Na+/HFD and Normo-Na+/HFD. On the other hand, SIK2 has shown to inhibits the expression of lipogenic genes in adipocytes [42]. The expression of SIK2 was significantly lower in High-Na+/HFD compared to Low-Na+/HFD. SIK2 has shown to regulates fatty acid oxidation, in the liver, skeletal muscle, and adipocytes [41].The expression of SGK1 is upregulated by several types of cell stress, it up-regulates Na+/K+-ATPase, amino-acid transporters, and several glucose carriers such as Na + coupled glucose transporter SGLT1. Thus, SGK1 supports cellular glucose uptake and glycolysis, angiogenesis, cell survival, cell migration, and wound healing family [43,44], we observed there was a downregulation of SGK1 in mice fed with High-Na+/HFD and an upregulation in those fed with Low-Na+/HFD.
It is important to note that in our experimental setting we did not potentiate fructose consumption by adding it into the drinking water. High fructose intake per se has been shown to induce NAFLD activating hepatic lipogenesis and inducing insulin resistance. Likewise, a report from Lanaspa et al. [45] showed that high salt consumption induced leptin resistance and obesity—and in consequence NAFLD—in a mechanism mediated by fructose metabolism. However, this experimental setting differs substantially since the source of sodium was high fructose, high salt in drinking water and the mechanisms related to feeding behavior are not the same as drinking. Interestingly, this is not the only report showing a paradoxical beneficial effect of high salt intake. Cui et al. showed that total fat mass and especially abdominal fat mass was decreased in mice fed with a high salt diet [38]. The proposed mechanisms behind involve the renin-angiotensin system, calcium signaling, and the participation of CREB1. However, the involvement of SIK1 and 2 was not addressed. In this way, our results suggest novel mechanisms insights to be addressed in further investigations. We show that high salt intake not only reduces steatosis in a setting where obesity and insulin resistance plays a central role as observed in the data obtained from HFD fed mice. But also in an experimental setting where these pathological features are virtually absent how occurs in the MCD mice model, suggesting that the mechanisms behind acts in an independent way. HFD and MCD mice model although different share the histological hallmarks observed in NAFLD patients and this is important because not all NAFLD patients are obese. Hence, our results could be extrapolated to the phenotype observed in lean NASH [46]. In the Fig. 6G, we summarized the results obtained and we proposed a plausible mechanism.
The main limitation of this study is the duration of the treatment probably representing an acute change in MR rather than chronic regarding high-Na + intake. It is anticipated that a long-term model will modify RAAS and MR levels differently. Additionally, these findings did not allow us to attribute causality and it was only evaluated for prevention and not for reversion of steatosis. However, these results warrant more studies to assess sodium and hepatic MR modulation. The clinical implications of our findings are not to promote a high-Na + diet but to encourage the potential use of MR antagonists early in the disease and the identification of downstream targets such as SIK1, SIK2 or SGK1. In conclusion, this study identified in two experimental models of NAFLD that MR inactivation can prevent steatohepatitis.AbbreviationsALT Alanine aminotransferase Acetyl-CoA carboxylase conjugated linoleic acid fatty acids Fatty acid synthase high fat diet Homeostasis Model Assessment-Insulin resistance insulin resistance lysophosphatidylcholine monocytes chemotaxic protein 1 metabolic syndrome mineralocorticoid receptor sodium nonalcoholic fatty liver disease Phosphatidylcholine phosphatidylethanolamine Phosphatidylinositol Phosphatidylserine renin angiotensin aldosterone system Stearoyl-CoA desaturase 1 serum-and-glucocorticoid-inducible kinase Salt-inducible kinase Sterol regulatory element-binding protein 1 Tissue inhibitor of Metalloproteinase 1 Tumoral necrosis factor-alpha
DC, IR, FR, NS, MP, MF, DSD, CAR, NT, JL, AR, FB, RB, PA, MA, JPA contributed to the conception and design of the study, acquisition of data, and analysis and interpretation of data, DC, IR, FR, CAR, AR, FB, RB, PA, MA, JPA contributed to drafting the article or revising it critically for important intellectual content, all authors approved the final version to be submitted.
Financial support/acknowledgmentsThis work was funded, in part, by grants from the Chilean Government [FONDECYT #1150327 and #1191145 to M.A.; #1200227 to JPA; #1190419 to R.B and #1191183 to F.B.; #1211879 to D.C.) and the Comisión Nacional de Investigación Científica y Tecnológica (CONICYT, AFB170005, CARE Chile UC)]. MA is part of the European- Latin American ESCALON consortium funded by the European Union’s Horizon 2020 Research and Innovation Program under grant agreement no. 825510. Funding from Ayudas para apoyar grupos de investigación del sistema Universitario Vasco (IT971-16 to P.A.), MCIU/AEI/FEDER, UE (RTI2018-095134-B-100 to P.A) is also acknowledged.
Conflict of interestThe authors do not declare any conflict of interest.
The following is Supplementary data to this article:



