



Primary biliary cholangitis and primary sclerosing cholangitis are rare diseases affecting the bile ducts and the liver. The limited knowledge of their pathogenesis leads to limited therapeutic options. Nevertheless, the landscape of novel therapies for these cholangiopathies is now rapidly changing, providing new treatment opportunities for patients and clinicians involved in their care. The aim of this review is to summarize the evidence of novel molecules under investigation for primary biliary cholangitis and primary sclerosing cholangitis and to discuss how they can potentially change current treatment paradigms.
Cholestatic liver diseases represent an heterogenous group of genetic and non-genetic conditions characterized by different pathogenesis, clinical features and prognosis [1]. This review will focus on primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC), two complex diseases of the bile ducts with immune-mediated etiopathogenesis requiring highly specialized care. For decades clinicians have had very few treatment options, while now many new molecules are investigated in clinical trials.
The aim of this work is to provide the state-of-the-art of novel therapies for primary biliary cholangitis and primary sclerosing cholangitis, and to analyze how new molecules might change therapeutic management of these diseases.
2Novel therapies in PBCPBC is an autoimmune cholestatic liver disease characterized by a chronic inflammation of the small bile ducts which, if not properly treated, leads to the destruction of the intrahepatic biliary tree, culminating in end-stage biliary cirrhosis [2].
The current therapeutic approach consists of two steps. The first one is to give all patients ursodeoxycholic acid (UDCA) at the dose of 13–15mg/kg/day. The second one is to evaluate after twelve months of stable treatment with UDCA the degree of biochemical response [3].
From 20% to 40% of the patients with PBC have an inadequate response to UDCA after twelve months, and the variance in the percentages mostly depend on the different definitions of biochemical response [3]. Since 2016, patients with alkaline phosphatase (ALP) greater than 1.5 times the upper limit of normal (ULN) after twelve months of UDCA may benefit from a second-line treatment, obeticholic acid (OCA). Nevertheless, nearly 50% of patients do not respond to the combination of UDCA and OCA [4]; in addition, the main adverse effect of OCA is pruritus, which is also one of the main symptoms of PBC [3,4]. It turns out there is still a relevant fraction of patients with PBC who are in need for novel treatments.
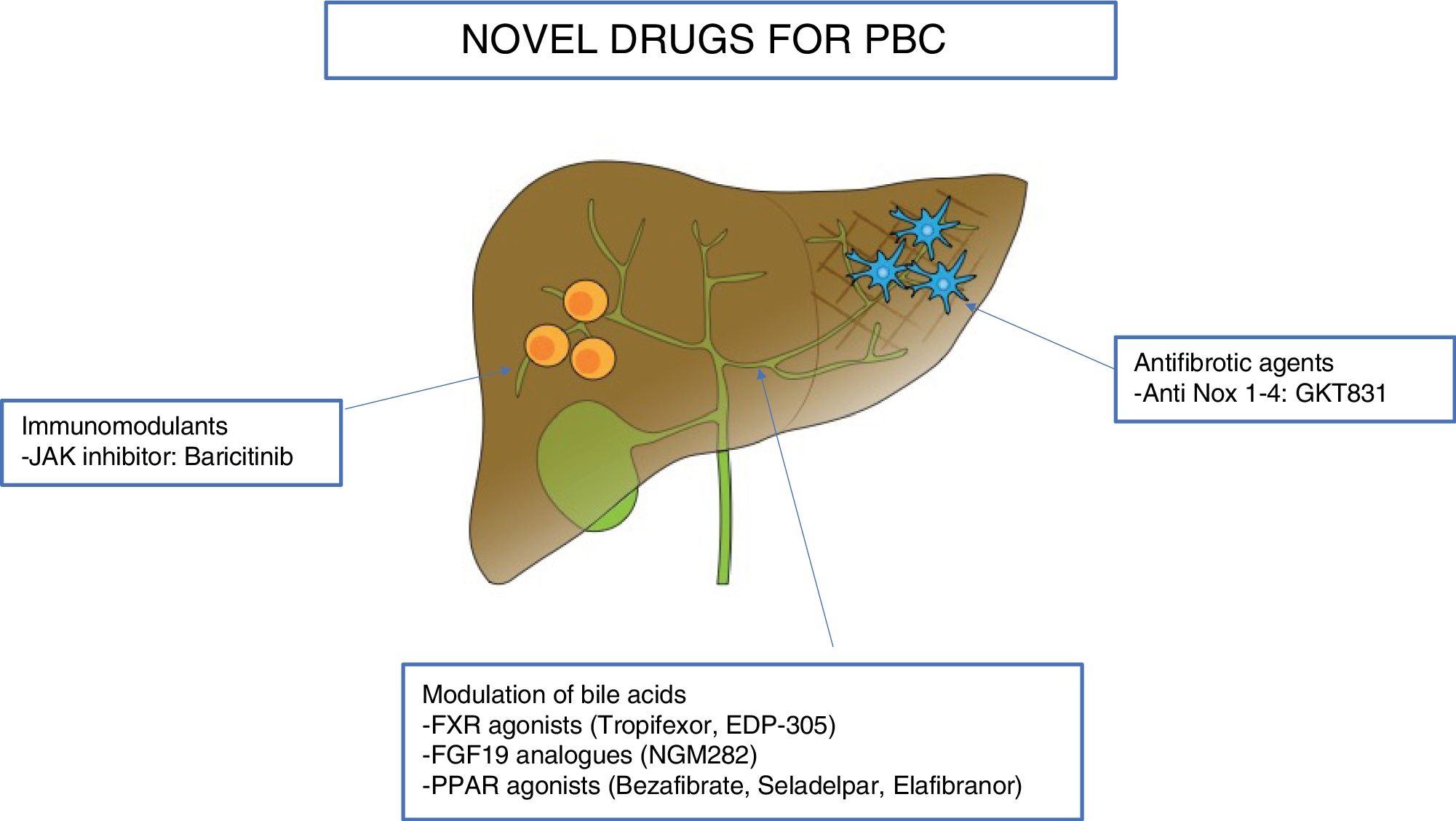
In this section we will review the novel drugs for the treatment of PBC, divided according to their target: modulation of bile acids, immunomodulatory agents, and antifibrotic molecules. Table 1 and Fig. 1 summarize the novel therapies investigated in PBC.
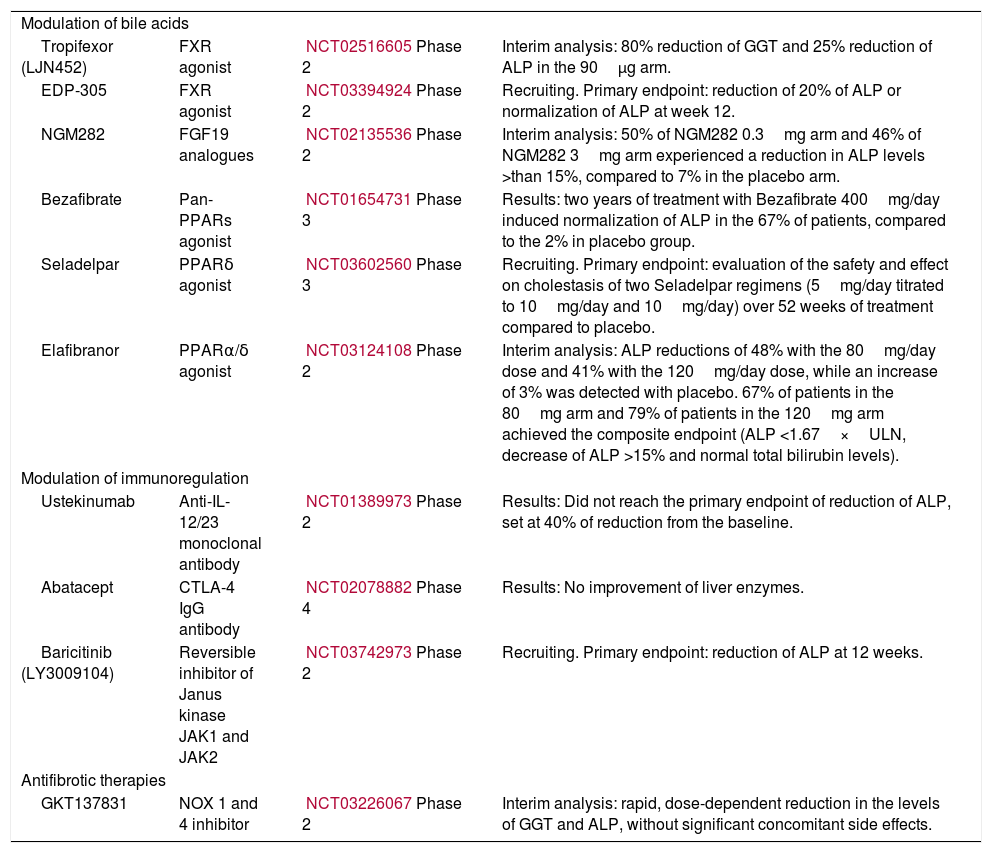
Novel therapies in primary biliary cholangitis.
| Modulation of bile acids | |||
| Tropifexor (LJN452) | FXR agonist | NCT02516605Phase 2 | Interim analysis: 80% reduction of GGT and 25% reduction of ALP in the 90μg arm. |
| EDP-305 | FXR agonist | NCT03394924Phase 2 | Recruiting. Primary endpoint: reduction of 20% of ALP or normalization of ALP at week 12. |
| NGM282 | FGF19 analogues | NCT02135536Phase 2 | Interim analysis: 50% of NGM282 0.3mg arm and 46% of NGM282 3mg arm experienced a reduction in ALP levels >than 15%, compared to 7% in the placebo arm. |
| Bezafibrate | Pan-PPARs agonist | NCT01654731Phase 3 | Results: two years of treatment with Bezafibrate 400mg/day induced normalization of ALP in the 67% of patients, compared to the 2% in placebo group. |
| Seladelpar | PPARδ agonist | NCT03602560Phase 3 | Recruiting. Primary endpoint: evaluation of the safety and effect on cholestasis of two Seladelpar regimens (5mg/day titrated to 10mg/day and 10mg/day) over 52 weeks of treatment compared to placebo. |
| Elafibranor | PPARα/δ agonist | NCT03124108Phase 2 | Interim analysis: ALP reductions of 48% with the 80mg/day dose and 41% with the 120mg/day dose, while an increase of 3% was detected with placebo. 67% of patients in the 80mg arm and 79% of patients in the 120mg arm achieved the composite endpoint (ALP <1.67×ULN, decrease of ALP >15% and normal total bilirubin levels). |
| Modulation of immunoregulation | |||
| Ustekinumab | Anti-IL-12/23 monoclonal antibody | NCT01389973Phase 2 | Results: Did not reach the primary endpoint of reduction of ALP, set at 40% of reduction from the baseline. |
| Abatacept | CTLA-4 IgG antibody | NCT02078882Phase 4 | Results: No improvement of liver enzymes. |
| Baricitinib (LY3009104) | Reversible inhibitor of Janus kinase JAK1 and JAK2 | NCT03742973Phase 2 | Recruiting. Primary endpoint: reduction of ALP at 12 weeks. |
| Antifibrotic therapies | |||
| GKT137831 | NOX 1 and 4 inhibitor | NCT03226067Phase 2 | Interim analysis: rapid, dose-dependent reduction in the levels of GGT and ALP, without significant concomitant side effects. |
. FXR, farnesoid X receptor; FGF19, fibroblast growth factor 19; PPAR, peroxisome proliferator-activated receptor.")
Farnesoid X Receptor (FXR) is a bile acid receptor expressed in the liver and in the small intestine. The activation of FXR regulates the enterohepatic bile acid homeostasis, inflammation, and fibrosis in response to liver injury [5].
OCA is a semi-synthetic bile acid acting as a FXR ligand, with a 100-fold greater potency in activating FXR than the natural analogue bile acid [6]. The safety and efficacy of OCA has been evaluated in the POISE study, a twelve-month, double-blind, randomized, placebo-controlled, phase III trial, in which the primary endpoint was the achievement of alkaline ALP <1.67×ULN, together with reduction of ≥15% from baseline and normal total bilirubin concentrations. The primary endpoint occurred in 46% and 47% of patients in the 5–10mg and 10mg OCA group, respectively, and 10% in the placebo group [4]. In line with these positive results, international guidelines now recommend OCA as first-line therapy in patients intolerant to UDCA, or as second-line therapy in addition to UDCA in patients with an incomplete response to UDCA [3,7,8]. Clinicians should start with 5mg/day and up-titrate to 10mg/day in case of non-response after six months.
The three-year interim data from the ongoing five-year POISE open-label extension study, evaluating the long-term efficacy and safety of OCA in PBC, have been recently published [9]. The reduction of ALP, bilirubin, gamma-glutamyl transferase (GGT), alanine transaminase (ALT) and aspartate transaminase (AST) serum concentrations were maintained up to 48 months of therapy with OCA. 48 months of OCA produce a predicted reduction in the risk of liver-transplantation or death when evaluated by the GLOBE score [10] or the UK-PBC risk score [11]. Regarding safety of OCA, liver-related and cardiovascular events, incidence of pruritus, and standard lipid analysis were evaluated throughout the double-blind and open-label phases. Pruritus represented the main adverse event, occurring in 77% of patients, of whom 4% discontinued treatment while 29% received a symptomatic therapy with a bile acid sequestrant. Concerning the lipid profile, during the open-label extension study an initial increase and a following large decrease of LDL cholesterol was observed. A decreasing trend for HDL, VLDL, total cholesterol and triglycerides was also detected.
Unfortunately, OCA does not represent the clinical answer for all patients with PBC for several reasons. Nearly half of patients treated with OCA will not achieve biochemical response [4]. Itching, which is a common issue in PBC, deeply affecting quality of life and functioning, is potentially worsened by OCA. Lack of longitudinal histological data do not allow solid conclusions about the ability of OCA to interfere with all the biological components of PBC. For example, in OCA-responders we have no data whether ductopenia, which is a feature of severe PBC [12], is affected by the treatment; likewise, it is still to be proven the capacity of OCA to reduce, or even revert, biliary fibrosis. It turns out that further studies on OCA are needed, together with the development of other molecules.
Tropifexor (LJN452) is a non-bile acid FXR agonist investigated as a potential new therapy in PBC [13]. Being a non-steroidal compound, it should not cause alterations in the lipid profile, differently from OCA. Safety, tolerability and efficacy of three doses of Tropifexor (30μg, 60μg and 90μg) have been evaluated in a double-blind, randomized, placebo-controlled, phase 2 study. Patients were included if they had ALP >1.67×ULN after twelve months of stable therapy with UDCA. Interestingly, the primary endpoint of this study was the reduction of GGT levels instead of ALP, to avoid the potential confounding effect of FXR-mediated ALP gene induction. Four-week interim analysis showed a rapid, dose-dependent reduction of GGT and ALP levels, with nearly 80% reduction of GGT and around 25% reduction of ALP in the 90μg arm. Transient changes in total, HDL and LDL cholesterol were observed in the four weeks of treatment, but levels returned to baseline by the end of the observational time of twelve weeks. Tropifexor proved to be safe and tolerable at 30–90mcg doses, without any serious adverse events, including discontinuations due to itch or transaminitis [14].
These unpublished data require further investigation, as safety on longer treatment schedule needs to be assessed. Moreover, while ALP is a validated prognostic and predictive biomarker in PBC, it is still to be ascertained whether reduction of GGT levels translates into prevention of liver-related outcomes (ClinicalTrials.gov Identifier: NCT02516605).
EDP-305 is another novel FXR receptor agonist which reduced the progression of fibrosis in animal models [15]. It is now under study in a randomized, double-blind, placebo-controlled phase 2 trial which will assess safety, tolerability, pharmacokinetics and efficacy in patients with PBC and inadequate response or intolerant to UDCA. Treatment schedule will be twelve weeks treatment with EDP-305 or placebo, and the primary endpoint will be the reduction of 20% of ALP or normalization of ALP at week twelve (ClinicalTrials.gov Identifier: NCT03394924).
2.1.2Fibroblast growth factor 19 analoguesFibroblast growth factor (FGF)19 is a hormone induced in the gut by FXR, and acts in the liver by suppressing CYP7A1, the enzyme that catalyses the first rate-limiting step in the classic pathway of bile acids synthesis [5,16,17]. Since FGF19 has been found to increase the risk of liver cancer in animal models, a synthetic analogue NGM282, was developed [18]. NGM282 regulates bile acids synthesis like FGF19 but does not have a carcinogenic potential. A 28-day, double-blind, placebo-controlled, phase 2 trial evaluated safety and efficacy of two formulation of NGM282 (0.3mg and 3mg) in patients with PBC and incomplete response after twelve months of stable therapy with UDCA (defined as ALP >1.67×ULN). Fifty percent of those receiving NGM282 0.3mg and 46% of those receiving NGM282 3mg experienced a drop in ALP levels greater than 15%, compared to 7% in the placebo group. Indeed, the kinetic of ALP drop was quick, and ALT, AST, GGT, immunoglobulin G (IgG), immunoglobulin M (IgM) and total cholesterol levels also improved. The observed decrease in complement component 4 (C4) levels in both treatment groups suggest that NGM282 acts by directly inhibiting the de-novo bile acid synthesis through the classical pathway. NGM282 was safe and well-tolerated, the main adverse effect being diarrhoea, and it did not induce or worsen pruritus [19] (ClinicalTrials.gov Identifier NCT02135536).
This is the first trial to report the efficacy of FGF19 analogues in patients with PBC, but this study has some limitations worth mentioning. Compared to other phase 2 trials in patients with PBC the duration of the study was much shorter, preventing firm conclusions about the long-term efficacy and safety of this molecule. It should also be noticed that NGM282 is administered subcutaneously, which might be less convenient than oral route and could potentially affect compliance.
2.1.3Peroxisome proliferator-activated receptors agonistsPeroxisome proliferator-activated receptors (PPARs) are nuclear receptors regulating the transcription of several genes involved in metabolic pathways and inflammation. PPARα is expressed in different tissues of the body, including the liver, and its action is to negatively regulate inflammatory pathways. The physiology of PPARα in the liver has been recently reviewed in detail elsewhere [20]. PPARβ/δ is specifically expressed in hepatocytes, cholangiocytes, Kupffer cells, hepatic stellate cells, and its activation in these cells seems to play a role in the progression of PBC [21,22]. Cholangiocytes use PPARδ to regulate transporters to absorb and secrete bile components [22]. PPARδ also has anti-inflammatory effects in macrophages, and anti-fibrotic effects in Kupffer and stellate cells [23]. On this basis, different PPAR agonists have been investigated as a potential therapy for PBC.
Bezafibrate (pan-PPAR agonist) has shown promising activity in decreasing cholestasis in PBC patients, acting on the downregulation of genes involved in bile acid synthesis and transport. Bezafibrate was evaluated in the BEZURSO trial. This landmark trial was a 24 months, double-blind, randomized, placebo-controlled, phase 3 trial, comparing the combination of UDCA and bezafibrate 400mg to UDCA and placebo in 100 patients with PBC and incomplete response after twelve months of UDCA treatment, defined according to Paris 2 Criteria [24]. The primary outcome of this study was a composite of normalization of ALP, AST, ALT, total bilirubin, albumin and INR at 24 months and was met by 31% of patients compared to 0% in placebo arm. Two years of treatment with Bezafibrate 400mg induced normalization of ALP in the 67% of patients, compared to the 2% in placebo group. Bezafibrate did not worsen pruritus, which was even improved in a subset of cases. Non-invasive markers of fibrosis decreased significantly in the treated arm (−15%, compared to +22% in placebo arm), although no improvements in disease activity and staging were demonstrated in the 28 patients with available longitudinal histological data [25] (ClinicalTrials.gov Identifier NCT01654731).
Safety issues have been raised regarding bezafibrate. In the BEZURSO trial there were no statistical differences regarding overall adverse events and serious adverse events between the two groups. However, well-known side effects of fibrates, like myalgias, transaminitis and increase in creatine kinase and creatinine did occur more frequently in those treated with Bezafibrate. Authors suggested that Bezafibrate should be used with caution in patients at risk for chronic kidney disease (e.g. those with diabetes or hypertension) or established renal disease [25]. Similarly, the possible negative impact on the underlying liver disease of hepatitis during bezafibrate therapy is still to be determined.
The BEZURSO trial had the merit of a long treatment schedule (two years) and of striking results in terms of biochemical response (achieving normalization in a considerable proportion of subjects), together with a significant activity on pruritus. It would be possible that also other drugs under investigation might arise safety concerns when treatment was prolonged more than four–twelve weeks. Conversely, patients included had lower values of ALP compared to other trials (e.g. POISE), and this might partially account for the higher rates of full biochemical response. Around half of the cohort was composed by advanced patients, which can be considered a plus of the study; nonetheless, these were those less likely to benefit, as post hoc analyses clearly showed (portal hypertension was independently associated with treatment failure). The lack of improvement in histological measures is also a matter of concern; prospective studies evaluating hard endpoints are needed.
Fenofibrate is a PPARα agonist and most of experience derive from small cohorts [26–30]. No randomized controlled trial are available and therefore its use is not encouraged, since data on efficacy are not sound and safety issues are reasonably the same of Bezafibrate, which has been studied in a solid randomized, placebo-controlled trial.
Seladelpar is a new selective agonist of the PPARδ receptor. A double-blind, randomized, placebo-controlled, phase 2, proof-of-concept study evaluated for the first time the anti-cholestatic effects and safety of Seladelpar in patients with inadequate response to UDCA (defined as ALP >1.67×ULN after twelve months of stable treatment) [31]. The primary endpoint was safety of two formulations, Seladelpar 50mg and Seladelpar 200mg, on a twelve-week treatment schedule. Unfortunately, the study was terminated early because of the occurrence of grade 3 transaminitis in three subjects. Interestingly, those subjects who completed twelve weeks of treatment fully normalized ALP. A 8-week, dose-ranging, open-label, randomized, Phase 2 study with a 44-week extension evaluated safety and efficacy of lower doses of Seladelpar (2mg, 5mg and 10mg) in PBC patients with incomplete response to UDCA, is ongoing (ClinicalTrials.gov Identifier: NCT02955602). Finally, the ENHANCE trial, a 52-week, double-blind, placebo-controlled, randomized, Phase 3 study that will evaluate the safety and efficacy of Seladelpar, is also actively recruiting. It includes subjects with PBC and an inadequate response to or intolerance to UDCA (here defined as ALP >1.67×ULN at twelve months of treatment) that will be randomized to a titration arm (5–10mg), 10mg arm or placebo (ClinicalTrials.gov Identifier: NCT03602560).
The efficacy and safety of Elafibranor, a dual PPARα/δ agonist, has been recently tested in a multicentre, randomized, double-blind, placebo-controlled, phase 2 study, evaluating Elafibranor 80mg and 120mg vs placebo. Patients were recruited if diagnosed with PBC and showing inadequate response to UDCA after twelve months of treatment (ALP >1.67×ULN); the primary endpoint of the study was the percentage change from baseline in ALP at week twelve compared to UDCA monotherapy. Unpublished data have been presented at the International Liver Congress in Vienna in April 2019 [32]. Both treatment arms experienced a significant decrease of the mean ALP compared to placebo; reductions of 48% with the 80mg/day dose and 41% with the 120mg/day dose were observed, while an increase of 3% was detected with placebo. 67% of patients in the 80mg arm and 79% of patients in the 120mg arm achieved the composite endpoint (ALP <1.67×ULN, decrease of ALP >15% and normal total bilirubin levels). Further significant improvements in lipid, inflammatory markers and pruritus were observed. Additional studies are needed to confirm these promising results (ClinicalTrials.gov Identifier: NCT03124108).
2.2Immunomodulatory agentsThe breach of tolerance due to immunoregulatory failure is considered the initial mechanism involved in the pathogenesis of PBC [33]. However, whether PBC is a “classical” autoimmune condition is still matter of debate, since immunosuppressive drugs seem not effective [34–39] or, like budesonide [40–42], it is highly debatable whether the trade-off between efficacy and safety is in favour of efficacy. Moreover, several biologics have been disappointing in PBC.
Rituximab is an anti-CD20 antibody commonly used to treat a broad range of lymphomas and autoimmune diseases thanks to its ability to deplete B-cells. B-cell depletion strategies have shown efficacy in murine models of autoimmune cholangitis and a number of clinical trials have been performed [43]. In an open label study on incomplete responders to UDCA, two doses of 1000mg of Rituximab determined a significant reduction in the titre of autoantibodies and IgM levels and no safety issues arose. The primary endpoint of the trial was safety but a little reduction of ALP at 36 weeks was noticed, despite no changes in other liver enzymes occurred [44]. A similar study was performed on 16 patients not responding to therapeutic doses of UDCA and being given two doses of 1000mg of Rituximab. The primary endpoint, normalization or improvement in ALP levels of at least 25% from baseline, was not reached, achieving a reduction of only 16% [44]. A phase 2, randomized, placebo-controlled trial on Rituximab in PBC has recently been published, but the primary endpoint was improvement in fatigue assessed by PBC-40 questionnaire. The trial failed to reach its goal, but some complementary analysis on ALP values did show a little improvement. It is important to underline that in this study only patients with early disease were allowed, and most of them were UDCA-responders [45]. Genome-wide association studies (GWAS) have identified a few single-nucleotide polymorphisms (SNPs) which are more frequently associated with the PBC phenotype; the identified loci may be pathogenetically relevant and their downstream biological pathways potentially druggable [46].
Ustekinumab is an anti-interleukin (IL)-12/23 monoclonal antibody currently used in treatment of several autoimmune conditions like psoriasis [47,48] and inflammatory bowel diseases (IBD) [49]. After a promising signal on IL-12 locus came up from a pioneer North-American GWAS study [50], a multicentre, open label trial was developed with the aim of blocking this pathway by Ustekinumab. Unfortunately, this multicentre open label proof-of-concept study did not reach the primary endpoint of reduction of ALP, set at 40% of reduction from the baseline [51].
T-cell interfering strategies have subsequently been investigated, supported by some preclinical evidence [52]. A key player in T-cell activation is Cytotoxic T-Lymphocyte Antigen 4 which binds to CD80 and CD86 on antigen-presenting cells hampering the costimulatory signal for the T-cell lymphocyte. Abatacept is a Cytotoxic T-Lymphocyte Antigen 4 IgG antibody currently used in clinical practice in rheumatoid arthritis [53] and psoriatic arthritis [54]. Abatacept was evaluated in PBC in an open-label trial where the drug was administered for 24 weeks. No major side effects occurred and Abatacept was effective in blunting activation of T cells; unfortunately, there was no improvement of biochemical enzymes [39] (ClinicalTrials.gov Identifier: NCT02078882).
Despite these discouraging studies other immunomodulants are under investigation in PBC. All the drugs described so far belong to the class of monoclonal antibodies; nonetheless, small molecules are also approaching the field of immune-mediated diseases [55], after having been used for years in oncology [56] and haematology [57].
Baricitinib (LY3009104) is a reversible inhibitor of Janus kinase JAK1 and JAK2. JAK–STAT pathway is exploited by many cytokines involved in the pathogenesis of autoimmune diseases [55], and Baricitinib is currently used in clinical practice as a disease-modifying drug for rheumatoid arthritis [55]. A randomized double-blind, placebo-controlled, phase 2, proof of concept study is currently ongoing to evaluate the efficacy and safety of Baricitinib in patients with PBC and incomplete response to UDCA (ClinicalTrials.gov Identifier: NCT03742973).
The previous failure of immunomodulant strategies in PBC might partly be explained by a temporal issue in the design of the trials, i.e. drugs given too late in the disease course, when cholestasis predominates over immunological abnormalities [39]. Moreover, it might be possible that biochemical enzymes are not capable to detect what occur in the liver, and longitudinal biopsies might be a valuable tool in future trials evaluating immunomodulatory drugs.
2.3Antifibrotic therapiesThe vicious cycle of inflammation, cholestasis and scarring characterize the pathological sequence from normal liver tissue to cirrhosis [58]. In cholestatic liver diseases, biliary fibrosis starts from portal tracts and progressively extends towards the lobule, leading to biliary-type cirrhosis [59].
The inflammation of small bile ducts is the main driver of liver damage in PBC; both the licensed therapies and the most promising agents under evaluation have a composite effect, being intrinsically choleretic but also carrying anti-inflammatory properties [3]. Nonetheless, to act also on the fibrogenic pathway and, possibly, reverting established fibrosis would be innovative.
Among the pro-fibrogenic molecules in cholestatic diseases, there is solid evidence supporting the role of reactive species of oxygen [59]. The Nicotinamide Adenine Dinucleotide Phosphate (NADPH) oxidases enzymes generate reactive species of oxygen, and play a physiological role under acute stress. However, chronic stress induces an excessive activation contributing to the inflammation-fibrosis process [58,60]. NADPH oxidases amplify a broad range of signalling pathways involved in inflammatory and fibrotic disorders across multiple organs and diseases [61].
GKT137831 is a first-in-class small molecule which acts through inhibition of NADPH oxidases isoforms 1 and 4 and exerts antinflammatory and antifibrotic effects. In animal models of acute biliary injury (bile-duct ligation) and steatohepatitis GKT137831 attenuated liver fibrosis, decreased hepatocyte apoptosis and reactive species of oxygen production [62,63]. Clinical trials evaluating GKT137831 in patients with diabetic nephropathy have demonstrated an excellent safety profile. GKT137831 is the first potential non-anticholestatic compound significantly improving markers of cholestasis and inflammation in PBC. A multicentre, randomized, double-blind, placebo-controlled, phase 2 study evaluating safety and efficacy of GKT137831 in patients with PBC and incomplete response has recently terminated the recruitment phase. A six-week ad-interim analysis has shown a rapid, dose-dependent reduction in the levels of GGT and ALP, without significant concomitant side effects [64] (ClinicalTrials.gov Identifier: NCT03226067).
2.4Future perspectives in PBCThe clinical scenario is constellated of several new potential therapies for PBC, driven by the increasing knowledge of the pathophysiological mechanisms of the disease. From the introduction of UDCA, the natural history of PBC has dramatically changed [65], despite stable trends in liver transplants for sicker cases [66]. The future challenge would be to treat every patient with a tailored therapy [67]. Nevertheless, there is need to define which endpoints we should look at, the timing of second-line therapy and which potential combinations of molecules are the safest and most effective ones.
The evidence supporting ALP as surrogate endpoint is solid and the decline of its levels is typically employed as treatment endpoint [10,11,68]. However, as previously outlined, in two recent clinical trials GGT was used as primary endpoint instead of ALP. Indeed, GGT is a cholestatic marker along with ALP [69], but has never been considered before, probably due to its low specificity. GGT levels can be elevated in patients with dysmetabolic conditions, but it is worth mention that also ALP levels can be influenced by other conditions. For example, osteomalacia is often associated to cholestatic liver diseases like PBC and can raise ALP due to the increase of bone fraction of ALP [70]. To us, the lack of evidence behind the use of GGT as treatment endpoint should prompt further investigation on the potential role of GGT as prognostic factor and potential treatment endpoint.
Another critical point in evaluating treatment efficacy is that we commonly lack longitudinal histological data. Despite in most cases liver biopsy is not necessary to diagnose PBC, it would provide information on the biology of the disease, which may not always be represented by the available serum biomarkers. Whether new molecules positively change some biological components of PBC like bile duct loss could only be speculated, since no data are available.
As far as timing of second-line treatment is concerned, we suggest that the current stepwise therapeutic approach might be hazardous for high-risk patients, in that they have to wait at least twelve months to establish treatment failure and receive a second-line drug. To improve this aspect, a predictive UDCA-response score was developed [71], and further research is needed to validate it. The aim would be to apply this score in clinical practice to reduce the time lag in which patients remain with an uncontrolled disease.
Furthermore, a debate on the management of patients not responders to OCA has also started after a Belgian study evaluated the effects of “triple” therapy with UDCA, OCA and bezafibrate. Eleven patients were selected from the POISE cohort, their main feature being persistently abnormal ALP despite five years of treatment with UDCA and titrated OCA. These subjects were started on bezafibrate treatment 400mg for six months and 44% of them achieved normalization of ALP. Moreover, a statistically significant decrease of ALP was observed in 100% of patients, along with an improvement of pruritus [72]. Caution is needed, since these are unpublished data and need further validation, but they did stimulate the scientific community to start delineating novel algorithms for the treatment of PBC. The acquirement of the license to develop and commercialize bezafibrate in the United States by Intercept with the aim to start a Phase 2 study of a combination with OCA is likely to offer new data in the close future [73].
3Part 2: novel therapies in PSCPSC is a rare and complex disease affecting large and small bile ducts, characterized by a slow and difficult-to-predict disease course towards biliary cirrhosis and death. It is considered a pre-cancerous condition, since the risk of cholangiocarcinoma and gallbladder cancer is extremely high in affected patients. The only effective treatment is liver transplantation, since no pharmacological measure is licensed. UDCA, the mainstay of therapy in PBC, can improve cholestatic enzymes in patients with PSC when used at doses of 13–25mg/kg, but clinical trials have failed to prove a survival benefit. Conflicting indications are present in current guidelines, so that UDCA treatment in PSC is still controversial [74].
Two main issues have contributed to the lack of therapies in PSC: the incomplete comprehension of the pathogenesis and the absence of reliable surrogate endpoints of outcome.
Despite the efforts to elucidate the pathogenesis, we are far from having a full understanding of mechanisms underlying PSC. It is still debatable whether the Mdr2−/− model is a valid animal model for PSC, since it does not develop inflammatory bowel disease and cholangiocarcinoma, while developing inflammatory-driven hepatocellular carcinoma [75].
The long and erratic natural history of PSC makes the use of hard endpoints (survival, need for transplantation) hardly feasible. Hence, in previous and ongoing clinical trials, reduction in levels of ALP is commonly used as surrogate endpoint. While in PBC the role of ALP has been validated in different cohorts [10,11], similar studies in PSC are still not available, even though there have been recent efforts to characterize ALP variability to improve the design of future trials [76]. Moreover, fluctuations of ALP during episodes of bacterial cholangitis, which often occur in PSC, complicate its use as a predictor of therapeutic response.
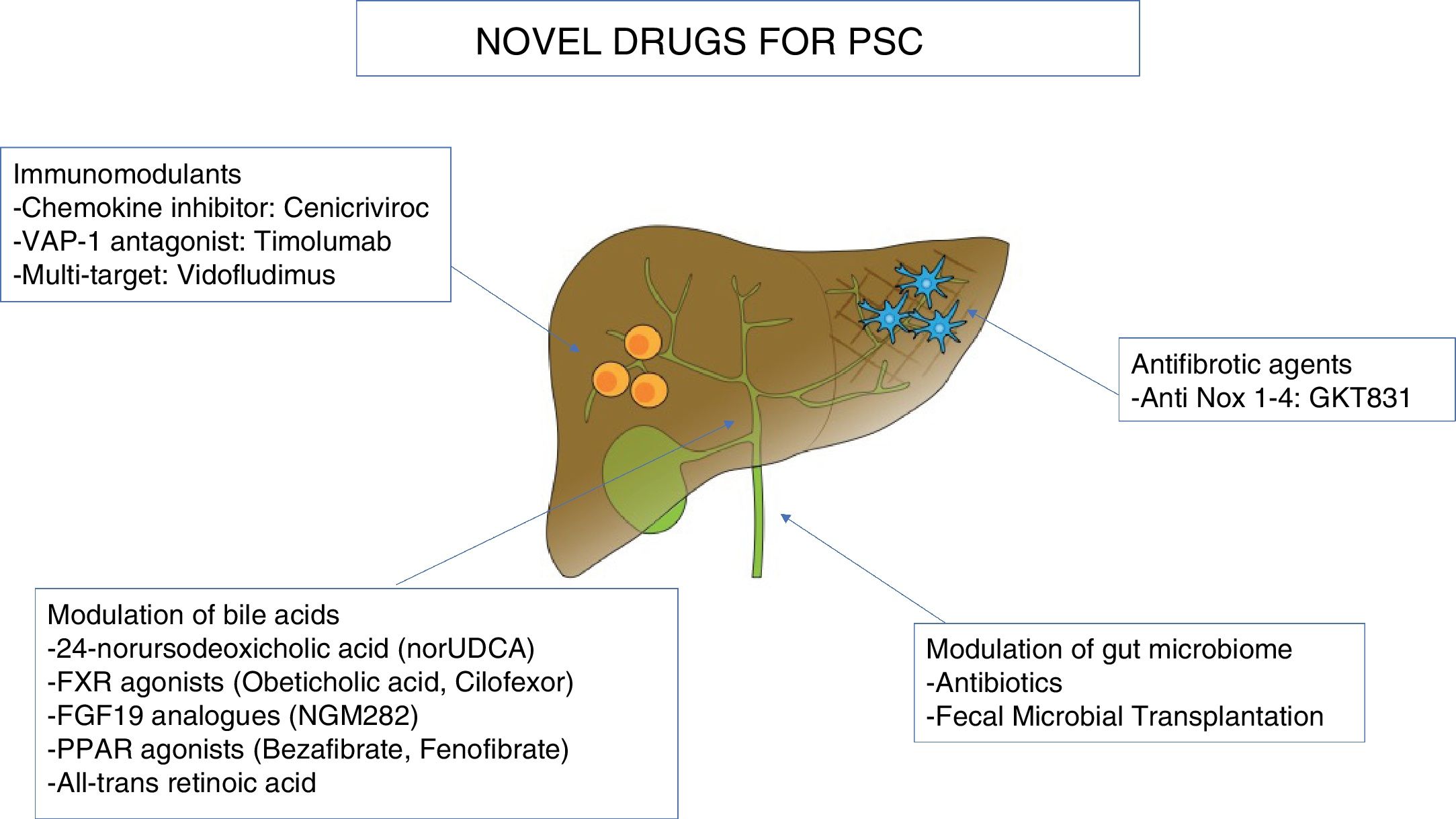
In this section, we will review the ongoing clinical trials in PSC, pooling together novel agents along three major pathogenetic theories: modulation of bile acids, immunomodulants, and change of the microbiome. Moreover, due to the marked fibrotic deposition in PSC, antifibrotic agents under investigation will be outlined. Table 2 and Fig. 2 summarize the novel molecules investigated in PSC.
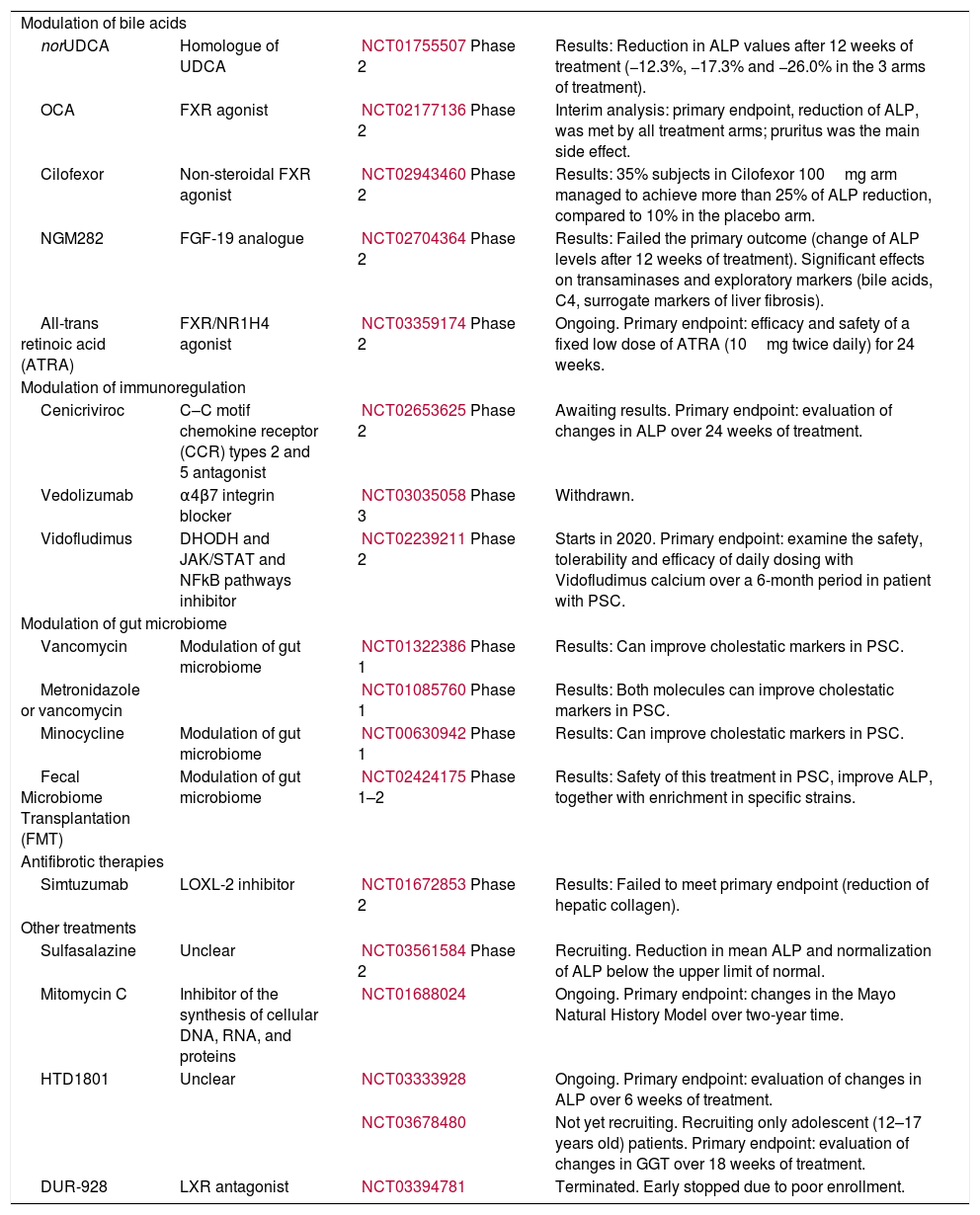
Novel therapies in primary sclerosing cholangitis.
| Modulation of bile acids | |||
| norUDCA | Homologue of UDCA | NCT01755507Phase 2 | Results: Reduction in ALP values after 12 weeks of treatment (−12.3%, −17.3% and −26.0% in the 3 arms of treatment). |
| OCA | FXR agonist | NCT02177136Phase 2 | Interim analysis: primary endpoint, reduction of ALP, was met by all treatment arms; pruritus was the main side effect. |
| Cilofexor | Non-steroidal FXR agonist | NCT02943460Phase 2 | Results: 35% subjects in Cilofexor 100mg arm managed to achieve more than 25% of ALP reduction, compared to 10% in the placebo arm. |
| NGM282 | FGF-19 analogue | NCT02704364Phase 2 | Results: Failed the primary outcome (change of ALP levels after 12 weeks of treatment). Significant effects on transaminases and exploratory markers (bile acids, C4, surrogate markers of liver fibrosis). |
| All-trans retinoic acid (ATRA) | FXR/NR1H4 agonist | NCT03359174Phase 2 | Ongoing. Primary endpoint: efficacy and safety of a fixed low dose of ATRA (10mg twice daily) for 24 weeks. |
| Modulation of immunoregulation | |||
| Cenicriviroc | C–C motif chemokine receptor (CCR) types 2 and 5 antagonist | NCT02653625Phase 2 | Awaiting results. Primary endpoint: evaluation of changes in ALP over 24 weeks of treatment. |
| Vedolizumab | α4β7 integrin blocker | NCT03035058Phase 3 | Withdrawn. |
| Vidofludimus | DHODH and JAK/STAT and NFkB pathways inhibitor | NCT02239211Phase 2 | Starts in 2020. Primary endpoint: examine the safety, tolerability and efficacy of daily dosing with Vidofludimus calcium over a 6-month period in patient with PSC. |
| Modulation of gut microbiome | |||
| Vancomycin | Modulation of gut microbiome | NCT01322386Phase 1 | Results: Can improve cholestatic markers in PSC. |
| Metronidazole or vancomycin | NCT01085760Phase 1 | Results: Both molecules can improve cholestatic markers in PSC. | |
| Minocycline | Modulation of gut microbiome | NCT00630942Phase 1 | Results: Can improve cholestatic markers in PSC. |
| Fecal Microbiome Transplantation (FMT) | Modulation of gut microbiome | NCT02424175Phase 1–2 | Results: Safety of this treatment in PSC, improve ALP, together with enrichment in specific strains. |
| Antifibrotic therapies | |||
| Simtuzumab | LOXL-2 inhibitor | NCT01672853Phase 2 | Results: Failed to meet primary endpoint (reduction of hepatic collagen). |
| Other treatments | |||
| Sulfasalazine | Unclear | NCT03561584Phase 2 | Recruiting. Reduction in mean ALP and normalization of ALP below the upper limit of normal. |
| Mitomycin C | Inhibitor of the synthesis of cellular DNA, RNA, and proteins | NCT01688024 | Ongoing. Primary endpoint: changes in the Mayo Natural History Model over two-year time. |
| HTD1801 | Unclear | NCT03333928 | Ongoing. Primary endpoint: evaluation of changes in ALP over 6 weeks of treatment. |
| NCT03678480 | Not yet recruiting. Recruiting only adolescent (12–17 years old) patients. Primary endpoint: evaluation of changes in GGT over 18 weeks of treatment. | ||
| DUR-928 | LXR antagonist | NCT03394781 | Terminated. Early stopped due to poor enrollment. |
. norUDCA, 24-norursodeoxicholic acid; FXR, farnesoid X receptor; FGF19, fibroblast growth factor 19; PPAR, peroxisome proliferator-activated receptor; VAP-1, vascular adhesion protein 1.")
24-norursodeoxycholic acid (norUDCA) is an homologue of UDCA which can resist conjugation with taurine or glycine thanks to its different chemical properties. It is secreted into bile and then, being unconjugated, is in turn absorbed by cholangiocytes, undergoing cholehepatic shunting. Through this mechanism, cholangiocytes are supraphysiologically stimulated to secrete bicarbonates. The final result is that bile ducts are flushed by a bicarbonate-rich bile and protected via the bicarbonate “umbrella” [77]. The solid evidence coming from preclinical models [77] and the excellent safety profile in phase I studies have led to a placebo-controlled, phase II clinical trial, showing a good safety profile (comparable to placebo) and a significant, dose-dependent reduction in ALP values after twelve weeks of treatment (−12.3%, −17.3% and −26.0% in the three arms of treatment) [78]. A phase III double-blind, randomized clinical trial is actively recruiting patients across several centres worldwide (ClinicalTrials.gov Identifier: NCT03872921).
3.1.1FXR agonistsFXR agonists can potentially delay progression of PSC through a multifactorial process. FXR activation, both via FGF-19 and directly acting on hepatocytes, can inhibit bile acid synthesis in the liver and counteract toxic effects of cholestasis [79]. Data for FXR agonists in PSC are available for OCA and Cilofexor.
OCA has been evaluated in PSC in the AESOP trial, a randomized, double-blind, placebo-controlled, phase II study. The primary endpoint was reduction of ALP; patients were randomized to receive either placebo, OCA 1.5mg or 5mg for 24 weeks. Patients in treatment arms could receive up to 3mg or 10mg, respectively, after twelve weeks. Primary endpoint was met by all treatment arms, while pruritus was the main side effect [80]. A phase 3 trial is awaited (ClinicalTrials.gov Identifier: NCT02177136).
Cilofexor is a non-steroidal FXR agonist that has been tested in a twelve-week, phase II, randomized, placebo-controlled clinical trial in patients with PSC without cirrhosis and ALP levels greater than 1.67× the ULN. Two doses (30mg and 100mg) were evaluated, and the primary endpoint was safety. Cilofexor proved to be safe and was also effective in reducing ALP levels together with GGT, ALT and AST. In particular, 35% subjects in Cilofexor 100mg arm managed to achieve more than 25% of ALP reduction, compared to 10% in the placebo arm. The main limitations of this study are the inclusion of only large-duct PSC cases, without cirrhosis and the low prevalence of IBD in their cohort [79] (ClinicalTrials.gov Identifier: NCT02943460).
The FGF19 analogue NGM282 has been recently tested in a phase II, randomized, double-blind, placebo-controlled study in patients with PSC and ALP levels greater than 1.5×ULN [81]. The study design allowed two doses of NGM282 (1mg or 3mg) to be administered for twelve weeks; the primary outcome was change of ALP levels after twelve weeks of treatment. This study has arisen much debate in the community, after failing the primary endpoint but showing significant reduction in other biologically-relevant markers such as bile acids, C4, transaminases and surrogate markers of liver fibrosis [81]. Indeed, this trial has awakened again the discussion around limitations of ALP as surrogate endpoint of novel therapies in PSC [81]. It is of note that this trial included also patients with dominant strictures, small-duct PSC, autoimmune hepatitis and cirrhosis, and this could have hampered the demonstration of the potential beneficial effects of the drug [82]. Despite it is probably likely that ALP is not the best treatment target, whether improvement of other explanatory markers translates into (1) improvement in hard outcomes, (2) prevention of cholangiocarcinoma needs also to be proven [82] (ClinicalTrials.gov Identifier: NCT02704364).
All-trans retinoic acid is a drug used for the treatment of acne and acute promyelocytic leukaemia, which is also capable to reduce bile acid pool size via the FXR/nuclear receptor subfamily 1, group H, member 4 (NR1H4) pathway [83]. It has shown favourable effects when combined with UDCA [84] or Cenicriviroc in preclinical models of cholestasis [85]. There is a single experience supporting some biological efficacy of the combination of UDCA and all-trans retinoic acid in patients with PSC [86]. Despite twelve-week of treatment did not reduce primary endpoint (ALP), the combination was able to reduce ALT and levels of C4 (i.e. to inhibit bile acid synthesis). An open-label trial evaluating efficacy and safety of a fixed low dose of all-trans retinoic acid (10mg twice daily) for 24 weeks is currently ongoing (ClinicalTrials.gov Identifier: NCT03359174).
3.1.2PPAR agonistsData about fibrates efficacy and safety in PSC are limited. No randomized controlled trials are available, and most of data come from retrospective case series, mainly from Japan [87,88]. Recently, the French-Spanish experience has been published, reporting data on 20 patients with PSC and persistently elevated ALP levels despite therapeutic doses of UDCA [89]. Authors reported around 40% reduction in ALP levels after three months of treatment with either fenofibrate (200mg/day) or bezafibrate (400mg/day). No safety concerns have arisen in their experience. The obvious limitations of a small, retrospective and uncontrolled study are evident; nonetheless, it would be interesting to investigate efficacy and safety of this class in a well-powered, randomized controlled trial. Indeed, the proven efficacy of bezafibrate in PBC [25] and the antinflammatory and anticholestatic effects deriving from PPAR agonism do support further studies.
3.2ImmunomodulantsPSC is considered an immune-mediated disease, as proven by clustering with other autoimmune conditions and recurrence after liver transplantation, but lacks specific autoantibodies and does not responding to traditional immunosuppressive approaches [74]. Previous trials with immunosuppressive drugs have failed to show a benefit in PSC [84]. Intriguingly, different molecules which target immune cells are showing encouraging results in PSC though.
Cenicriviroc is a novel drug antagonizing C–C motif chemokine receptor (CCR) types 2 and 5 which showed promising results in preclinical models of cholestasis (Bile-duct ligated rat and Mdr2−/− mouse) [85] and in patients with NASH [90]. Inflammatory cells (neutrophils and T cells) were reduced in the liver of treated BDL rats and Mdr2−/− mice. Interestingly, Cenicriviroc alone was inferior to a combination of Cenicriviroc and all-trans retinoic acid, suggesting the potentiality of multimodal approaches in PSC [85]. PERSEUS is an open label, proof of concept study in patients with PSC which aimed to evaluate changes in ALP over 24 weeks of treatment with Cenicriviroc. Results are still awaited (ClinicalTrials.gov Identifier: NCT02653625).
Other promising immunomodulants have been borrowed from the treatment armamentarium of IBD. The intimate connection between PSC and IBD is well established, despite disease courses do not go along [74]. Differently from PSC, effective treatments for IBD do exist, like Vedolizumab [91]. There is evidence that the targets of Vedolizumab (α4β7 integrin and mucosal addressin cellular adhesion molecule (1) belong to a pathway involved in the pathogenesis of PSC [92,93]. Leucocyte trafficking between the gut and the liver is one of the key component of the gut–liver axis model [94], and has been thoroughly explained elsewhere [95,96]. Despite these good premises, a phase III trial evaluating Vedolizumab for patients with both PSC and IBD has been retired in 2018 (ClinicalTrials.gov Identifier: NCT03035058) and retrospective experience of PSC-IBD patients treated with Vedolizumab for their IBD did not show any improvement in the biliary counterpart [97].
Vidofludimus, a small molecule which has shown some efficacy in IBD in a small clinical trial [98], is currently under study in PSC. Its action is multifaceted, blocking replication of activated T- and B-cells through Dihydroorotate dehydrogenase (DHODH) inhibition [99] and exerting anti-inflammatory effects by interfering with the JAK/signal transducer and activator of transcription (STAT) and nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB) pathways [100]. A phase II, open-label clinical trial aiming to examine the safety, tolerability and efficacy of daily dosing with vidofludimus calcium over a 6-month period in patient with PSC is due to start in 2020 (ClinicalTrials.gov Identifier: NCT03722576).
Chronic liver inflammation is responsible of immune cell mediated destruction of bile ducts. Vascular adhesion protein-1 (VAP-1) is a sialoglycoprotein expressed in the endothelium of the liver vessels which favours adhesion of lymphocytes [101]. Gut–liver axis theory supports the notion that primed lymphocytes from the gut move to the liver and recognize VAP-1 which acts as chemokine. Strategies antagonizing VAP-1 are expected to reduce liver inflammation and fibrosis.
Timolumab is a human monoclonal antibody which binds to VAP-1 counteracting its action. BUTEO is a single-arm, open-label, multi-centre trial evaluating safety and efficacy of Timolumab in patients with PSC [102] (ClinicalTrials.gov Identifier: NCT02239211).
3.3Modulation of gut microbiomeThe liver is anatomically and physiologically linked to the gut by receiving intestine-derived blood through the portal vein, which is rich in nutrients and gut-derived bacteria [103,104]. It turns out that gut microbiota and liver are also tightly connected, and abnormalities in its composition (so-called dysbiosis) have been associated to many chronic liver diseases, including PSC [105]. The microbiome (i.e. the whole set of genes of a specific microbiota) of PSC patients is different from healthy controls and IBD-only patients [104]. Interestingly, the presence or not of concomitant IBD does not seem to have an impact on this discrepancy [104]. It is still not clear whether dysbiosis is the cause of bile ducts abnormalities or it is secondary to the changes occurring in the gut mucosa during chronic biliary diseases.
The interest in the potential role of gut microbiota as a therapeutic target in PSC derives from the hypothesis that the vicious cycle of inflammation and fibrosis in the bile ducts might be driven by bacteria getting through an inflamed intestinal mucosa [106]. By modulating their composition this cycle could potentially be broken, in turn reducing inflammation and scarring of the bile ducts. In addition, a beneficial effect in PSC of modulation of gut microbiota might be an indirect proof-of-concept of the theoretical pathogenetic role of dysbiosis in PSC [107].
The simplest strategy to change the composition of gut bacteria is to administer antibiotics with known action on microbiota. Available data in PSC come from some prospective trials, namely three randomized controlled trials and two uncontrolled studies [107], including metronidazole [108], minocycline [109], vancomycin [108,110] or rifaximin [111]. Despite the evidence is small, there is a signal that vancomycin, metronidazole and minocycline can improve cholestatic markers in PSC. The reason why these drugs did show some effect while rifaximin did not is difficult to explain based on available data and further studies are warranted (ClinicalTrials.gov Identifier: NCT00630942, NCT01085760, NCT01322386).
The second and more promising strategy is Fecal Microbiome Transplantation (FMT). FMT is an established strategy for the treatment of recurrent infection from Clostridium Difficile [112]. To our knowledge, only one pilot study has been published, showing the safety of this treatment in PSC [113]. In this trial, FMT was capable to increase diversity and in some patients improve ALP, together with enrichment in specific strains. A multicentre study of FMT in PSC is awaited. ClinicalTrials.gov Identifier: NCT02424175.
A possible future strategy is to investigate whether gut virome (resident viral community) [114] and mycobiome (resident fungal community) [115] are also altered in PSC and might be targeted. Abnormalities have been found in IBD and alcoholic liver disease [107], and a recent French study showed the presence of fungal gut dysbiosis independent of IBD in patients with PSC [116]. It is likely that novel ecological approaches to handle the complexity of host–microorganism interactions will foster this field in the next future [117–119].
3.4Antifibrotic agentsThe presence of scarring in small and large ducts, causing strictures and dilatations, is a sine-qua-non condition to diagnose PSC. Biliary fibrosis in PSC has peculiar morphological aspects, being characterized by a typical, but often absent, “onion-skin” shape of the connective tissue around the injured bile ducts. Despite fibrosis is a central aspect of the disease, very few direct antifibrotic options have been developed. A Lysyl oxidase like-2 (LOXL2) inhibitor called Simtuzumab represented a promising molecule but failed to show a benefit on a phase 2 trial aimed to assess its efficacy in reducing hepatic collagen [120] (ClinicalTrials.gov Identifier: NCT01672853).
The preclinical evidence of efficacy in murine models of cholestasis and the previously mentioned good preliminary results in PBC might support the study of GKT137831 as a potential treatment for PSC.
3.5Other treatmentsA list of other treatment with unclear mode of action under investigation in PSC is reported in Table 2.
3.6Future perspectives in PSCPSC is still an orphan disease, with many aspects still to be elucidated and no drugs of proven efficacy. Previous failures should probably invoke caution in judging new molecules under investigation. The main limiting factor in PSC, compared to PBC, is the erratic disease course and the lack of accurate biomarkers able to track disease progression and stratify patients in different risk groups. We are still limited in evaluating whether a novel drug is really a disease-modifying agent in PSC. Although higher ALP values are often related to more severe cholestasis, so that its reduction might translate in some clinical benefit, its fluctuations hamper its role as reliable treatment endpoint [81]. An international effort should be established to validate a solid biomarker that can be used as reliable endpoint of clinical trials of novel molecules for PSC.
With these limitations in mind, failed trials have also included different treatment endpoints (e.g. simtuzumab) and it is also possible failure occurred because investigated molecules were merely ineffective. Nonetheless, the simtuzumab case is a good example that well-designed trials still represent a precious chance to have prospective data in PSC.
4Innovative therapeutic approaches for autoimmune cholangiopathiesA different approach to treat autoimmune liver diseases is the use of nanoparticles. There is evidence that nanoparticles could be much useful to induce tolerance in autoimmune diseases in general [121].
Promising data have been produced for autoimmune hepatitis in preclinical models by using Avidin-Nucleic-Acid-Nano-Assemblies (ANANAS) nanoparticles to carry dexamethasone selectively to the liver. These studies mainly focused on reducing the systemic exposure of steroids through a selective mean of delivery [122]. A more recent work from the University of Calgary, Canada has taken advantage of peptide-major histocompatibility complex class II (pMHCII)-based nanoparticles, where the peptide of choice was the mitochondrial pyruvate dehydrogenase complex-E2 component. After administering these nanomedicines to different animal models resembling PBC (NOD.c3c4 mice, ARE-Del−/− mice) they were able to show the differentiation of FoxP3−CD25− T-cells into T-regulatory-type-1 (TR1)-like cells, which in turn suppressed local autoantigen presentation and antigen-presenting cell activation. This ability of pMHCII-based nanoparticles to blunt autoimmunity has already been proven in animal models of type 1 diabetes, multiple sclerosis and rheumatoid arthritis [123]. Interestingly, it was of much interest that nanoparticles relevant for PBC (like PDC-E2 nanoparticles) were effective also when used in non-PBC animal models, like Abcb4 knockout mice (model resembling PSC) or Ad-hFTCD mice (model resembling AIH type 2). Furthermore, researchers were able to reveal that nanoparticles did not impair cellular or humoral immunity towards foreign antigens [124]. The novelty behind these findings is the evidence that liver autoimmunity is probably antagonized in an organ-specific rather than a disease-specific manner, and this might translate in the future use of a few nanomedicines for different autoimmune liver diseases. Hopefully, nanoparticle-based approaches could help to entangle the complexity of the treatment of immune-mediated cholangiopathies and their safety and efficacy can be tested in clinical trials soon.
5ConclusionsIn this review we have outlined several molecules which can play a role in the therapeutic armamentarium of PBC and PSC. Although, we agree that cholestatic liver diseases will not be properly treated until a multimodal approach is employed [125]. Pathogenesis of PBC and PSC include dysregulation of the immune system, inflammation, bile acid toxicity and fibrogenesis, while current treatment strategies are limited to only one or two of these components. In other diseases, like rheumatoid arthritis, a multimodal approach is already part of clinical practice [126]. Although, it should be noted that this assumption is probably more valid for PBC, while data to support it in PSC are lacking, even though theoretically it is also reasonable [127].
Nonetheless, multimodality is not the only key to succeed. We need to understand the key drivers of these diseases also in relation to the time factor, i.e. when a specific dysregulation plays a major role and when its role is taken over by another one. To this end, we should move towards the development of targeted therapies to be used at the right stage. This would allow to combine really effective compounds, rather than just associating molecules with “cosmetic” effects on liver enzymes.
To us, one of the most promising and fruitful strategy to achieve this result is to combine a solid pathogenetic theory, taking advantage of the amount of experimental data generated by decades of hypothesis-driven studies in PBC and PSC, together with a deep sequencing of affected individuals by using next-generation sequencing techniques [128]. We speculate that precision medicine in cholestatic liver diseases will mean to profile patients at different -omic levels and then combine different treatments tailored to the results [128,129]. Multi-omics strategies (from genomics to metabolomics, going through metagenomics) [130], are already a reality in cancer medicine [131–133], diabetes [134] and neurodegenerative diseases [135], and should become part of practice also in hepatology, and in cholestatic liver diseases in particular [67,136–138].AbbreviationsPBC primary biliary cholangitis primary sclerosing cholangitis ursodeoxycholic acid alkaline phosphatase upper limit of normal obeticholic acid Farnesoid X Receptor gamma-glutamyl transferase alanine transaminase aspartate transaminase low-density lipoprotein high-density lipoprotein fibroblast growth factor immunoglobulin G immunoglobulin M complement component 4 peroxisome proliferator-activated receptor genome-wide association studies single-nucleotide polymorphism interleukin inflammatory bowel disease Janus kinase nicotinamide adenine dinucleotide phosphate 24-norursodeoxycholic acid nuclear receptor subfamily 1, group H, member 4 C–C motif chemokine receptor dihydroorotate dehydrogenase signal transducer and activator of transcription nuclear factor kappa-light-chain-enhancer of activated B cells vascular adhesion protein-1 fecal microbial transplantation lysyl oxidase like-2 Avidin-Nucleic-Acid-Nano-Assemblies peptide-major histocompatibility complex class II T-regulatory-type-1
Marco Carbone reports being on advisory committee and and consultant for Intercept; Pietro Invernizzi reports grant support from Intercept, Bruschettini and Gilead.
The authors of this publication are members of the European Reference Network on Hepatological Diseases (ERN RARE-LIVER). This work has been supported by grants of the Italian Ministry of Health in the role of auto-reactive hepatic natural killer cells in the pathogenesis of primary biliary cholangitis (PE-2016-02363915) and in the biocompatible nano-assemblies to increase the safety and the efficacy of steroid treatment against liver inflammation (GR-2018-12367794).



