




Lysosomal acid lipase deficiency (LAL-D) is an autosomal recessive disease caused by mutations in the LIPA gene, located on the long arm of chromosome 10 (10q23.31). Up until now, more than 59 mutations have been described and which are the cause of a very wide clinical spectrum. The goal of this study was to identify the mutations present in Mexican pediatric patients with a diagnosis of LAL-D.
Materials and methodsA cross-sectional study was carried out which included all the pediatric patients with LAL-D treated in a tertiary hospital in Mexico from January 2000 to June 2017.
ResultsSixteen patients with LAL-D were identified with a disease phenotype marked by the accumulation of cholesteryl esters. Eight distinct variants in the LIPA gene sequence were found, four pathogenic variants and four probably pathogenic. In six individuals, the variants were found in the homozygous state and ten were compound heterozygous. The eight variants were inverted, with five found on exon 4 and the others on exons 2, 8 and 10. The variant c.386A>G;p.His129Arg was the most common, being found in six of the 16 individuals (37.5%), making it much more frequent than what had previously been reported in the literature in proportion to the rest of the variants. The mutation known as E8SJM, which has been the mostly frequently found at the international level, was not the most common among this group of Mexican patients.
In conclusion, Mexican patients present a different frequency of mutations associated with LAL-D in comparison to European populations.
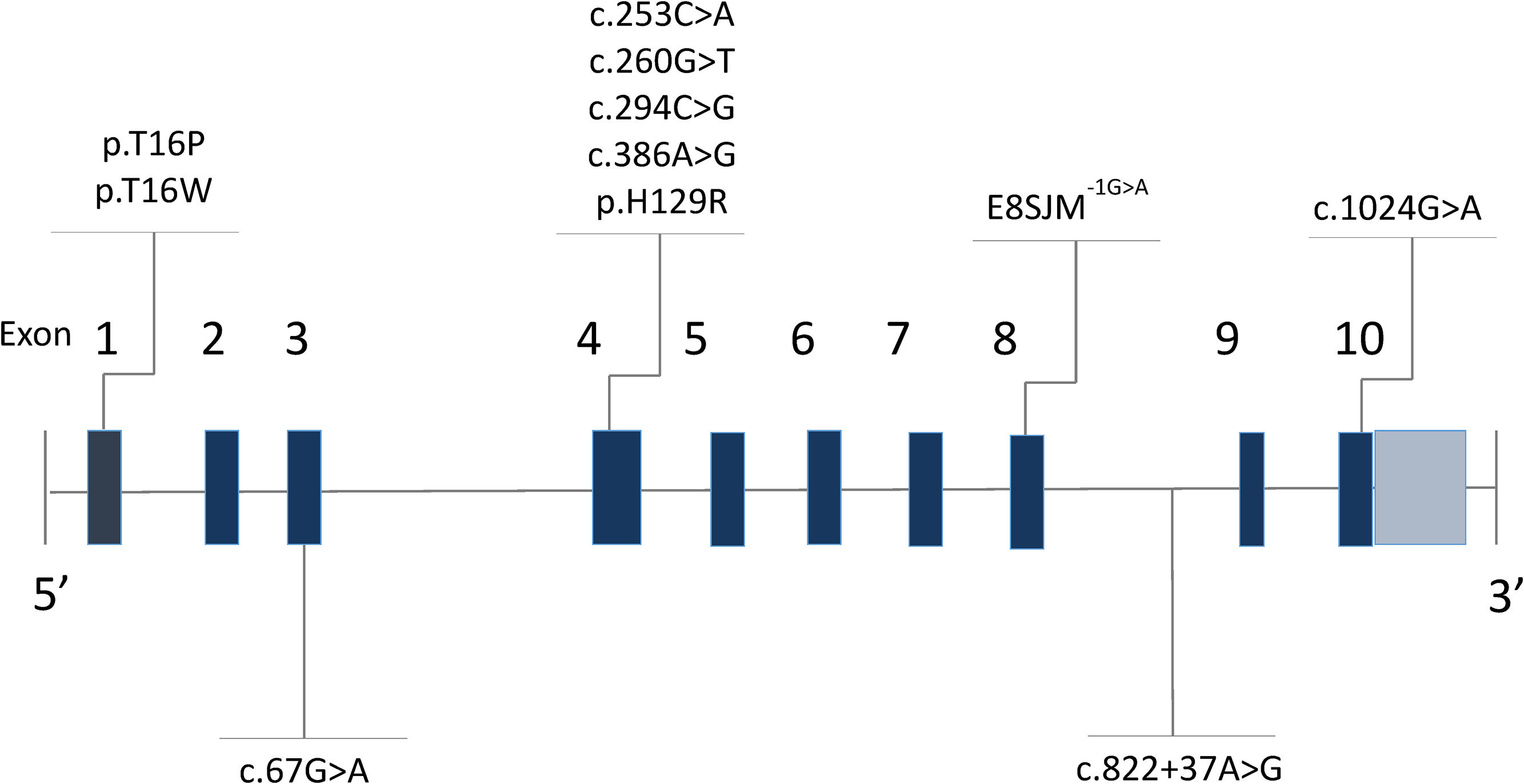
Lysosomal acid lipase deficiency (LAL-D) (MIM 278000) is an autosomal recessive disorder caused by mutations in the LIPA gene (MIM 613497) [1], located on the long arm of chromosome 10 (10q23.31) [2]. In 1994, Aslanidis and collaborators [2] described that the gene was made up of 10 exons and measured 45kb [3,4]. Intron 3 is characteristically wider than the rest (Fig. 1) [5,6]. The gene codes for the lysosomal acid lipase (LAL) protein, responsible for the hydrolysis of cholesteryl esters and triglycerides that have been internalized by the cell through lipoprotein endocytosis [7]; the misfunctioning of this enzyme leads to an accumulation of cholesteryl esters and triglycerides in the hepatocytes, intestine, adrenal glands and in the cells of the monocyte–macrophage system [8].

Up until now, more than 59 gene mutations have been described, which together are the cause of a wide clinical spectrum (Human Gene Mutation Database, Institute of Medical Genetics at the University of Cardiff, www.hgmd.cf.ac.uk). A recurring pathogenic variant in which there is a substitution of guanine for adenine in position 894 of cDNA (c.894G>A) has been reported within European populations with a frequency of around 60% [9]. This protein-level alteration would not produce a change in the amino acid (p.Gln298Gln) due to redundancy in the genetic code; nevertheless, mRNA is generated before the synthesis of amino acid, in which the substitution of guanine for adenine modifies the splicing of exon 8, leading to the deletion of 24 amino acids (p.delS275_Q298) [10]. This alteration is known as E8SJM-1G>A (Exon 8 Splice Junction Mutation), and this protein-level change can be found in the literature as either p.Gln298Gln or p.delS275_Q298, without distinction [11]. There have also been described small deletions/insertions, inverted mutations, being less common than large deletions and directionless mutations [12].
Clinically, LAL-D results in two main phenotypes: Wolman disease (WD) (MIM 278000), with early onset in the neonatal stage, and which corresponds to a fulminant subtype of the disease; and cholesteryl ester storage disease (CESD), which normally has a later onset, although it may present itself from infancy up to adulthood with a chronic evolution, but which without treatment may finally turn fatal [13,14]. It has been noted that when patients present a clinical picture beginning in the first months of life, it is due to directionless variants, such as in c.129C>G, which causes a codon for premature birth in position 43 of the amino acid (p,Tyr43*) [15] or in those that affect the reading frame (like c.414dup;p.Phe139llefs*7) [16], in contrast to those that manifest themselves clinically in the first decade of life or in adulthood, where the variants found have a lesser effect on the sequence of amino acids and produce an enzyme with certain residual activity (such as c.386A>G;p.His129Arg) [15]. However, despite these findings, no one has been able to definitively establish a clear phenotype-genotype relationship [11,17,18].
The goal of this study is to describe the mutations that are present in Mexican patients with a diagnosis of LAL-D.
2Materials and methodsThis is a descriptive study which included every patient with LAL-D treated at the Federico Gómez Children's Hospital of Mexico (HIMFG, in Spanish) from January 2000 to June 2017. DNA genomes were obtained from peripheral blood from all pediatric patients with a diagnosis of LAL-D that had been confirmed by enzyme activity while capillary sequencing of the LIPA gene was carried out by the Molecular Development Laboratory at University of Washington, Department of Pediatrics in Seattle, by the Haemetological and Metabolic Diseases Group Translational Research Unit, of the Instituto de Investigación Sanitaria (IIS) Aragón, GIIS-012 CIBER Rare diseases (CIBERER) or at Neurogenetics DNA Diagnostic
Laboratory, Massachusetts General Hospital, Boston, MA. All of the parents agreed to participate with their informed consent and patients eight years or older agreed to participate with their informed assent. Furthermore, the Research, Ethics, and Biosecurity Committees of the HIMFG gave their approval.
3ResultsSixteen patients with LAL-D were identified, eight of whom were male. The median age at the time of diagnosis was 6.3 years old with an RIC of 4. All of the patients manifested the cholesteryl ester accumulation variant of LAL-D, while none had presented early onset or Wolman disease. Eight distinct varieties in the LIPA gene sequence were found: four pathogenic variants (c.260G>T;p.Gly87Val, c.386A>G;p.His129Arg, c.894G>A;p.delS275_Q298, c.1024G>A;p.Gly342Arg) and four variants previously reported as probably pathogenic or of unknown effect (c.67G>A;p.Gly23Arg, c.253C>A;p.Gln85Lys, c.294C>G;p.Asn98Lys, c.894G>C; p.Gln298His). In six individuals, the variants were found in the homozygous state and ten were compound heterozygous. The eight variants were inverted, with four found on exon 4 and the others on exons 2, 8 and 10 (Table 1 and Fig. 1).
Variants of LIPA gene mutations found in the Mexican patients analyzed.
| Patient | Variant | Type | Change at the protein level | Effect/phenotype reporteda |
|---|---|---|---|---|
| 1 | c.260G>Tc.386A>Gc.67G>A | HeterozygousHeterozygousHeterozygous | p.Gly87Valp.His129Argp.Gly23Arg | Pathogenic/WD, CESDPathogenic/CESDUnknown effect/WD |
| 2 | c.386A>G | Homozygous | p.His129Arg | Pathogenic/CESD |
| 3a | c.253C>Ac.294C>G | HeterozygousHeterozygous | p.Gln85Lysp.Asn98Lys | Probably pathogenicProbably pathogenic |
| 4a | c.253C>Ac.294C>G | HeterozygousHeterozygous | p.Gly85Lysp.Asn98Lys | Probably pathogenicProbably pathogenic |
| 5 | c.253C>A | Homozygous | p.Gly85Lys | Probably pathogenic |
| 6 | c.894G>A | Homozygous | p.delS275_Q298 | Pathogenic/CESD |
| 7 | c.894G>A | Homozygous | p.delS275_Q298 | Pathogenic/CESD |
| 8 | c.1024G>Ac.894G>A | HeterozygousHeterozygous | p.Gly342Argp.delS275_Q298 | Pathogenic/CESDPathogenic/CESD |
| 9 | c.894G>A | Homozygous | p.delS275_Q298 | Pathogenic/CESD |
| 10 | c.67G>Ac.260G>T | HeterozygousHeterozygous | p.Gly23Argp.Gly87Val | Unknown effect/WDPathogenic/WD, CESD |
| 11 | c.386A>G | Homozygous | p.His129Arg | Pathogenic/CESD |
| 12 | c.386A>G | Homozygous | p.His129Arg | Pathogenic/CESD |
| 13 | c.253C>A | Homozygous | p.Gly85Lys | Probably pathogenic |
| 14 | c.386A>G | Homozygous | p.His129Arg | Pathogenic/CESD |
| 15 | c.67G>Ac.386A>G | HeterozygousHeterozygous | p.Gly23Argp.His129Arg | Unknown effect/WDPathogenic/CESD |
| 16 | c.894G>Ac.894G>C | HeterozygousHeterozygous | p.delSer275_Gln298p.Gln298His | Pathogenic/CESDProbably pathogenic/WD |
WD, Wolman disease; CESD, Cholesteryl Ester Storage Disease.
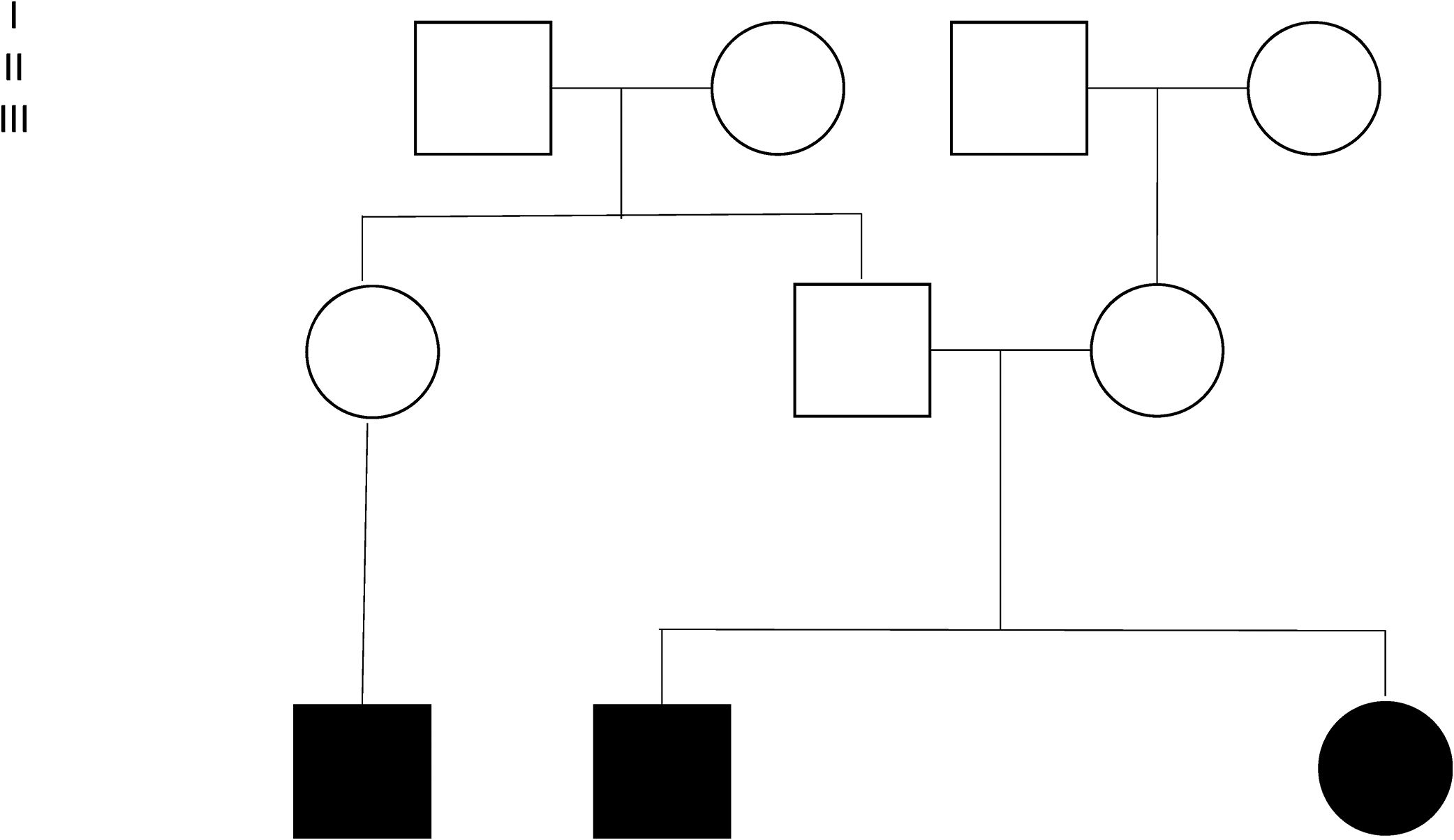
Four families were found among 15 of the individuals (Fig. 2). Individuals 3 and 4 are siblings from Mexico City, without antecedents of consanguinity. Two inverted variants were reported in them that produce a protein-level change (c.253C>A;p.Gln85Lys and c.294C>G;p.Asn98Lys) considered probably pathogenic. Individuals 5 and 13 are brothers belonging to a family from Villahermosa, Tabasco, also without antecedents of consanguinity. These patients were homozygous for the variant c.245C>A;pGly85Lys (reported as probably pathogenic). Individuals 6, 7 and 9 were members of the same family, heralding from Querétaro (Fig. 2). The pathogenic variant E8SJM-1G>A (c.894G>A; p.delS275_Q298) was found in these patients in the homozygous state. Family 4 was made up of individuals 8 and 16. Patient 16 is the mother of patient 8; the initial diagnosis was made in patient 8. Patient 16 had remained asymptomatic; however, as part of the preparation for the study of patients with a diagnosis of LAL-D, their parents’ enzyme activity was measured. Patient 16 was found to have diminished enzyme activity, as well as hepatomegaly and dyslipidemia; for those reasons it was decided to undertake a LIPA gene capillary sequencing. The study found that individuals 8 and 16 are compound heterozygote and that both share the pathogenic variant E8SJM-1G>A (c.894G>A;p.delS275_Q298) in the heterozygous state. Individual 8 additionally presents the pathogenic variant c.1024G>A; p.Gly342Arg in the heterozygous state, which demonstrated that it had been inherited from the father (Fig. 2). Furthermore, patient 16 presents the probably pathogenic variant c.894G>C;p.Gln298His in the heterozygous state.
4Discussion
This study describes 16 Mexican individuals with a diagnosis of LAL-D. It represents the first reported cohort of Mexican patients and the largest in Latin America. Eight distinct variants were detected in the 16 individuals analyzed. The patients hail from different states of the country; despite this, the variant c.386A>G;p.His129Arg was found in six individuals, which represents 37.5% of our population. It was found in the heterozygous state in two patients (individuals 1 and 15, Table 1) and in the homozygous state in four patients (individuals 2, 11, 12 and 14, Table 1). None of the six individuals belong to the same family or to nearby locations. This variant has been previously reported in the literature and has been designated pathogenic, especially related to the CESD clinical setting. The frequency of this variant in the general European population was given as 0.009 [15], which differs with what appears in this cohort of Mexican patients in which a much greater frequency is observed.
On the other hand, the variant E8SJM-1G>A (c.894G>A; p.delS275_Q298), which is more common in European populations, was only found in four individuals among our population, three of whom belong to the same family and presented it in the homozygous state. Scott and collaborators reported that the frequency of the variant E8SJM-1G>A (c.894G>A; p.delS275_Q298) depends on the ethnic group being studied, which could explain the lower frequency discovered in our population [19]. Due to the unequal distribution of this variant, directed screening is not currently recommended for this alteration in those individuals with a clinical suspicion of LAL-D, but rather a complete gene sequencing [11].
The variant c.67G>A;p.Gly23Arg was found in the heterozygous state in three patients, all of whom come from different regions of the country. This variant has been considered as probably benign by bioinformatic prediction (SIFT and PolyPhen); there are two reports, however, in international databases (www.ncbi.nlm.nih.gov), of two patients with this variant and Wolman disease. Due to this precedent and the three patients analyzed in this cohort who show a characteristic clinical picture for LAL-D, this could suggest that is a pathogenic variant.
Patients 3 and 4 were previously described by Santillán-Hernández and collaborators, and they currently remain in treatment at our institution. These two sisters are compound heterozygote (c.253C>A;p.Gly85Lys and c.294C>G; p.Asn98Lys), and the variants found had not been previously reported in the literature; nevertheless, through models that predict the protein-level alteration along with the low enzyme activity present, it was concluded that these variants are probably pathogenic [20]. This study describes another two siblings with the variant c.253C>A;p.Gly85Lys in the homozygous state with a medical history of LAL-D and low enzyme activity; when taken together with evidence from the literature, it suggests that this variant can be considered pathogenic.
The variant c.894G>C;pGln298His has previously been designated as probably pathogenic, a patient having been reported in an international database with clinical characteristics compatible with LAL-D (www.ncbi.nlm.nih.gov). The PolyPhen-2 prediction model reports it as probably harmful, while SIFT as harmful. Individual 16's report with the presence of this variant contributes evidence for considering it a pathogenic variant.
The rest of the variants found in this patient cohort (c.260G>T;p.Gly87Val, c.386A>G;p.His129Arg and c.1024G>A;p.Gly342Arg) had previously been reported in the literature and designated as pathogenic (Human Gene Mutation Database, Institute of Medical Genetics at the University of Cardiff, www.hgmd.cf.ac.uk).
This is the largest study of pediatric patients with LAL-D in Latin America that describes found mutations. Mexican patients present a different frequency of mutations associated with LAL-D in comparison to European populations. The report on patients with variations previously referred to as probably pathogenic or of unknown effect along with characteristic clinical manifestations of LAL-D provide evidence for considering them pathogenic. Given the limited number of patients, we could not conclude that there exists a correlation between the genotype and phenotype of the disease.AbbreviationsA adenine asparagine arginine cytosine complementary deoxyribonucleic acid cholesteryl ester storage disease deoxyribonucleic acid Exon 8 Splice Junction Mutation guanine glutamine glycine Hospital Infantil de México Federico Gómez (in spanish) histidine kilobase lysosomal acid lipase deficiency lipase A, lysosomal acid type lysine messenger ribonucleic acid phenylalanine polymorphism phenotyping glutamine interquartile range serine sorting intolerant from tolerant thymine tyrosine valine Wolman disease
There are no financial disclosures to declare.
Authors’ contributionsDr ACS and Dr RVF participated equally and sufficiently in the intellectual content, assembled the research protocol, contribute analyzing the data, write the paper, review the statistical analysis and approved the final manuscript as submitted.
Dr ARR contributes analyzing the data, writes the paper and approved the final manuscript as submitted.
Dr CPAB, MPOV and JJC contribute analyzing the evidence, and approved the final manuscript as submitted.
Conflicts of interestThe authors have no conflicts of interest to declare.
Informed patient consentAll of the parents agreed to participate with their informed consent and patients eight years or older agreed to participate with their informed assent, including publication of the case details.



