






Drug-induced hepatotoxicity is a significant and still unresolved clinical problem. The limitation in current knowledge regarding mechanisms of hepatic toxicity renders most of the preclinical review process failing and most of drug-induced hepatic injury remains unpredictable. Current knowledge on the mechanisms of drug-induced liver cell death is reviewed here. The intervention of both intra-and extracellular factors in determining the appearance of drug-induced cell apoptosis or necrosis is also discussed.
Finally, the role of both mitochondria and non parenchymal cells are reviewed with respect to approaches useful to manage drug-induced liver injury.
Cause of its location astride the primary port of entry for ingested substances and its richness of enzymes metabolizing xenobiotics, the liver is the main target for the toxicity of several compounds among which many medicaments.1 Most of drug-induced hepatic injuries are unpredictable and, although massive hepatic necrosis is a dramatic condition often requiring organ transplantation and account for an elevated number of fulminant hepatitis; luckily this event is rather infrequent if one considers the large consumption of medicaments. Another pathway potentially leading to liver damage goes through apoptosis; however, the distinction between apoptosis and necrosis may really appear idealized because both processes often coexist in the same microscopic field.2
Topics reviewed here deal with main mechanisms leading to drug-induced hepatic cell death and their relevance for prevention and treatment. Discussed are recent views on histology, apoptosis, necrosis, mitochondria, non-parenchymal cells, and immune response in the liver.
Histological evaluation of liver damageDifferent experimental methods used to study mechanisms of liver cell injury induced by toxic substances are reported in Table I. As shown, the suspected toxicity for a molecule can be experimentally tested by administering the compound at increasing doses, in the presence of metabolic inducers or inhibitors, with depletion of protective systems or with the use of toxicity enhancers.
Experimental methods to study drug hepatotoxicity.
| • Administration of the substance at increasing doses |
| • Use of metabolic inducers and inhibitors |
| • Depletion of protective systems |
| • Administration of toxicity enhancers (in the presence of low doses of the drug) |
| • Administration of the drug together with other known toxic substances |
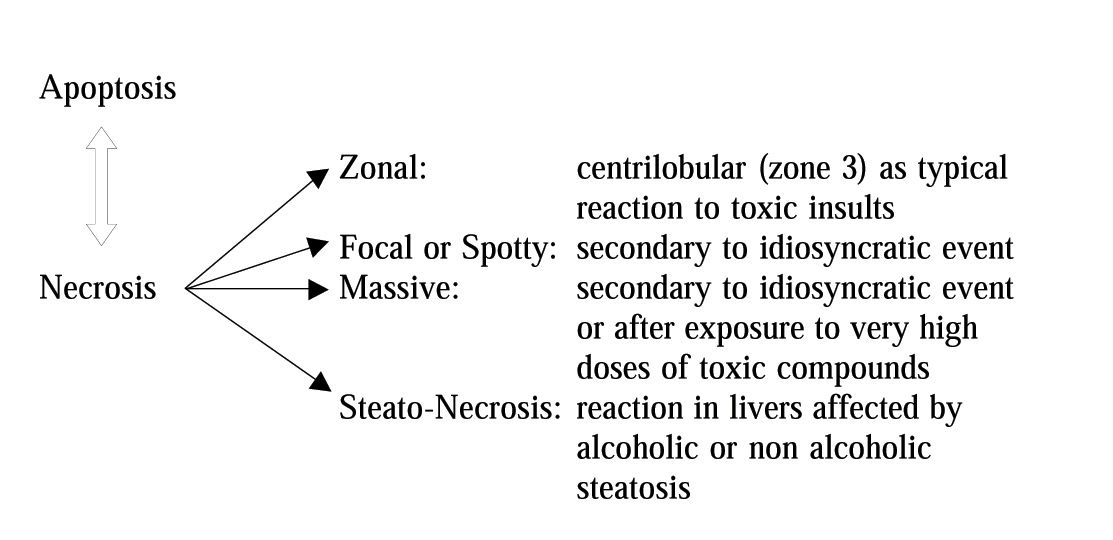
According to recent evidence, it seems that both apoptosis and necrosis may follow the activation of common or parallel metabolic pathways, thus rendering the differences more apparent than real.2 A typical example is the case of hepatic necrosis induced in zone 3 by low doses of carbon tetrachloride; whereas the administration of very low doses of the same compound is followed by the appearance of scattered single dead cells in zone 2 and 3, identified as apoptotic cells. However, it is current opinion that, in the presence of injuries incompatible with the maintenance of functional programs, hepatic cells preferentially die for apoptosis, because this death process does not obligatory activate an inflammatory response and may represent a protective mechanism for limiting the extent of the drug-induced liver damage. However, several factors interfere with the hepatocyte reaction to the insult coming from the drug, and the following development of necrosis or apoptosis may result from the sum of intrinsic and extrinsic factors to the cell.
It is generally accepted the distinction in a “direct toxic damage” which is dose dependent, predictable and experimentally reproducible and an “idiosyncratic damage” (metabolic or immune-mediated) which is not dose dependent, not predictable and not experimentally reproducible. Both these mechanisms of injury often show overlapping features identified as necrosis or apoptosis. In addition, a combination of environmental factors, hormones, nutritional and energetic status of the cell, and local oxygen tension, strongly contributes to the expression of cell death mediators.3
In the case of necrosis, the damage first appears at cytoplasmatic level; next, it spreads to the rest of the cell involving mitochondria and the nucleus, and finally results in swelling and loss of plasma membrane integrity leading to lysis. The necrotic process becomes irreversible when alterations of the ionic pump occur and result in increased cytosolic calcium concentration.4 The latter can depend on an increased release from intracellular organelles (mitochondria, endoplasmic reticulum) or an increased influx from outside environment. By contrast, apoptosis appears as cytoplasmic and nuclear condensation and fragmentation without loss of membrane integrity. Generally, drug-induced apoptosis is spotty, whereas necrosis tends to be zonal (Figure I).

However, whenever subjected to a toxic aggression, hepatocytes react by activating defense mechanisms or undergo irreversible damages; main putative mechanisms are shown in Table II. In other cases, hepatocyte injury may also follow the occlusion of centrolobular veins (azathioprin, estrogens, progesterone, pirrolidinic alkaloids) or follows severe vasoconstriction (cocaine).5
Hepatic cell response to toxic insults. Reactive response
| Reactive response |
| • Endoplasmic reticulum and monoxygenase hypertrophy |
| • Induction of protective systems (GSH) |
| • Synthesis of “heat shock” proteins |
| • Synthesis of acute phase proteins |
| Irreversible Response (cell death) |
| • Membrane alterations (Ca2+ influx) |
| • Mitochondrial swelling and decrease of energy production |
| • Progressive endoplasmic reticulum dilatation |
| • Cytoplasmic acidosis and DNA condensation |
Some of the mechanisms leading to cell apoptosis are well known and extensively studied, such as the p53 induced IL1B converting enzyme activation; conversely, the role played by many other molecules is still not well defined and is currently under investigations, such as the decrease of energy production and the activation of nucleases.6 However, it is commonly accepted the existence of an intrinsic and an extrinsic pathways of drug-induced hepatocyte apoptosis, which finally result in a common sequence of events.1
The intrinsic way is activated by a direct damage of the drug to nuclear and/or mitochondrial DNA with production of single-stranded DNA which subsequently stimulate an intracellular sensor system and induce the expression of p53. In the extrinsic way, the appearance of new surface antigens on the cell membrane induce the activation of cytotoxic T cells which release cytokines.7 Both these ways finally promote IL1B activation with subsequent involvement of caspases and nucleases. Accordingly, apoptosis can be initiated by intracellular events with activation of a cascade of reactions involving mitochondria,8 or by ligands, such as α-TNF (alpha-tumor necrosis factor) or FasL (Fas ligand), which engage death receptors on the cell surface.9,10 After the binding with the relative receptor, this trimerizes leading to a clustering of death domains.
Hepatocytes are generally resistant to α-TNF induced cytotoxicity.11 In fact, under normal condition, the activation of membrane receptors stimulates the synthesis of anti-apoptotic molecules (Bcl-2, NO-synthase) mediated by the intervention of the nuclear transduction factor (NF-kB). Therefore, an increased sensitivity of the cell to α-TNF-induced damage it is required, as it has been demonstrated in the case of ethanol, which requires a mitochondrial GSH depletion to sensitize the cell to α-TNF induced apoptosis.12
There are two major types of propagation of the apoptotic death program: a strong signaling response which directly activate the executioner caspases13 or, if the extent of the activation of caspase initiator is insufficient, other intracellular amplification mechanisms are required and usually involve mitochondria8(Figure 2).

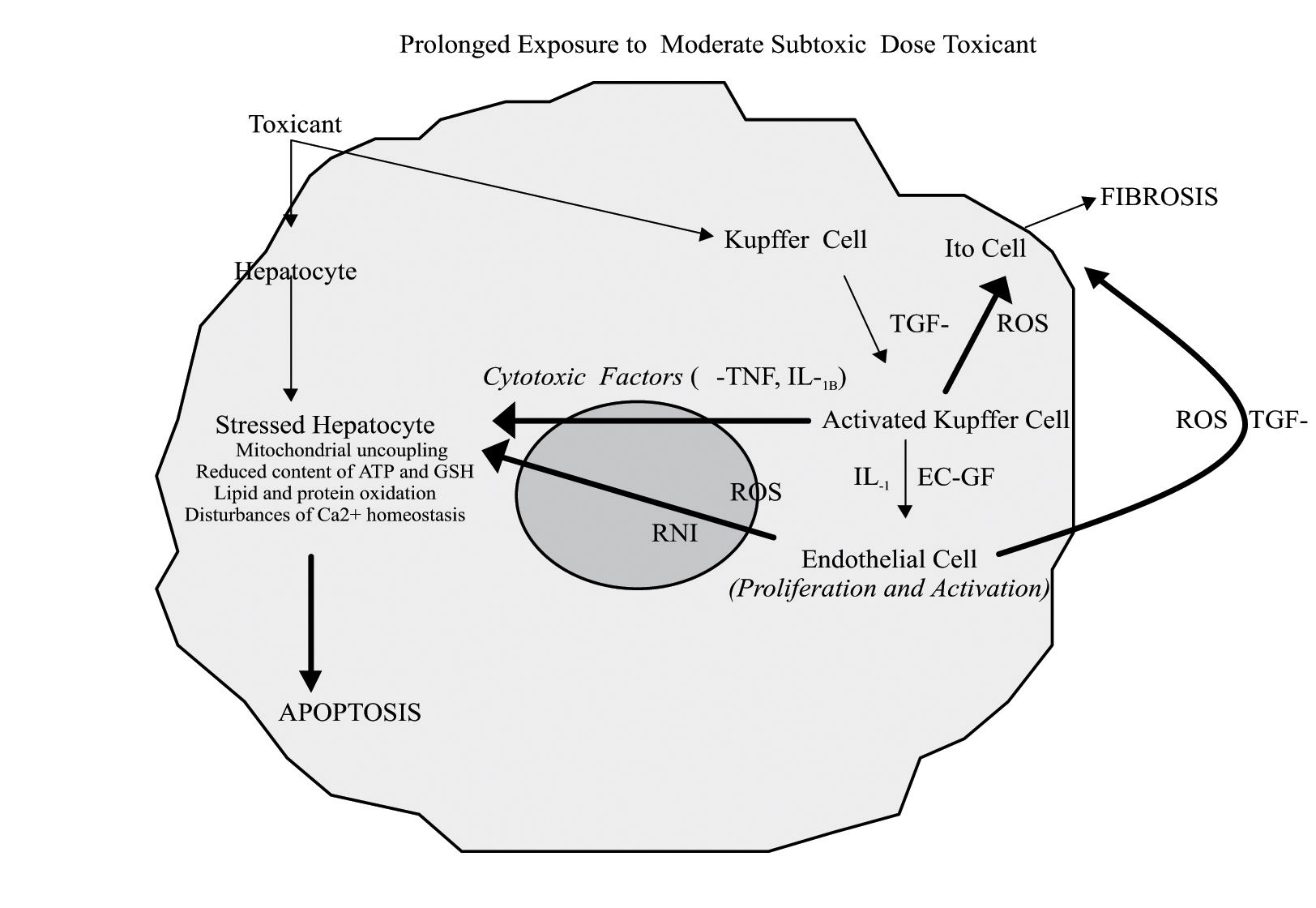
The participation of non parenchymal cells to drug-induced liver injury after exposure to prolonged subtoxic doses depends on the characteristics of the compound, dose, local oxygen tension, and other factors. In particular, α-TNF, released by Kupffer cells, induces apoptosis in sensitized hepatocytes showing mitochondrial uncoupling, reduced content of ATP or GSH, disturbance in calcium homeostasis, and which express α-TNF receptors on the membrane surface. EC-GF, endothelial cell growth factor; IL-1, interleukin-1; RNI, reactive nitrogen intermediates; ROS, reactive oxygen species; TGF-β, transforming growth factor beta.
Recent acquisitions on the pathogenesis of alcoholic liver injury indicate the participation of a high intestinal absorption of endotoxins and the following hepatocellular oxidative stress to the activation of Kupffer cells which, in turn, release pro-inflammatory cytokines (α-TNF, IL1B, IL6) and chemokines, thus playing an important role in ethanol-induced hepatic cell damage. Since it has been observed that the chemical elimination of Kupffer cells by Gadolinium chloride reduces also the extent of acetaminophen (APAP)-induced liver injury, it is likely that the activation of Kupffer cells may play a role also in APAP hepatotoxicity.14
NecrosisMany xenobiotics are lipophilic substances and are transformed in hydrophilic compounds after the cytochrome P-450 metabolic pass. Several factors affect the efficiency of the hepatic drug metabolic system: aging, liver disease, enzyme induction and inhibition, genetic (slow and fast acetylators), local oxygen tension.15 APAP is one of the most investigated drugs able to induce liver necrosis. When administered at low doses, APAP metabolites are mainly excreted after conjugation (90%) and oxidation (10%). At high doses, once the glucuronation and sulfatation pathways are saturated, excess of APAP is oxidized by the cytochrome P-450 enzymes with the release of large amount of the highly reactive metabolite NAPQI. This toxic metabolite stimulates lipid and protein oxidation both directly and after depletion of cytosolic and mitochondrial GSH through conjugation.16 It forms adducts with proteins which may have immunogenic properties and may activate Kupffer cells and polymor-phonuclear cells with subsequent release of free radicals. The oxidation of intracellular protein sulphydrils to form disulfide bonds results in an increased permeability of the inner mitochondrial membrane with consequent loss of potential, decrease in ATP synthesis, inhibition of Ca2+ dependent ATPase, reduced capability to sequester Ca2+ within mitochondria, actin oxidation with breakage of microfilaments and membrane blebs formation.17 Considered together, all these events are finally involved in the promotion of a necrotic cell death.
A threshold exists before compounds like APAP can determine the appearance of hepatic cell necrosis. It consists of the resultant between the production of reactive metabolites by the cytochrome P-450 and the ability of intracellular detoxifying systems and GSH in particular to scavenge them.18 In the presence of toxic metabolites, a reduced detoxification and/or excretion capacity by the cell may predispose to the injury. In particular, it has been shown that APAP associated necrosis occurs when hepatocellular concentrations of GSH fall below 1 μmol/g of hepatic tissue and mainly involves the perivenous level (zone 3). Therefore, conditions associated with a low hepatic reserve of detoxifying molecules, as in the case of ethanol abuse, may predispose hepatocytes to APAP or other medicaments-induced necrosis19,20(Figure 3).

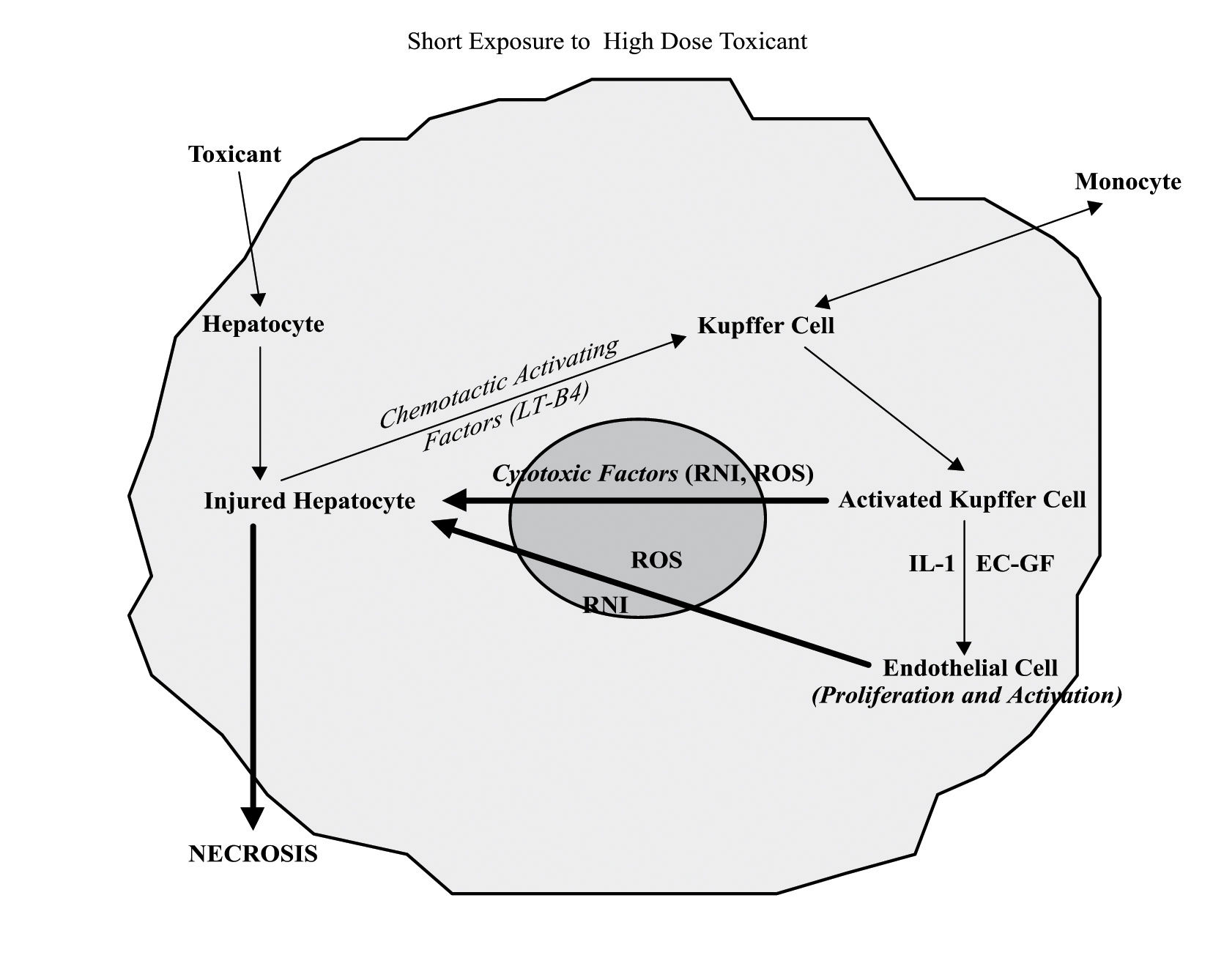
The participation of non parenchymal cells to liver cell damage induced by high dose of toxicants follows the release of chemotactic activating factors by injured hepatocytes. The activation of Kupffer and endothelial cells is associated with release of great amounts of free radicals and cytotoxic factors, which finally contribute to the hepatocyte necrotic death. EC-GF, endothelial cell growth factor; IL-1, interleukin-1; RNI, reactive nitrogen intermediates; ROS, reactive oxygen species; LTB4, leukotriene B4.
It has been recently shown that an alteration of the control mechanisms of cell volume is a factor promoting the appearance of hepatocyte necrosis. In the presence of massive oxidative stress, xenobiotic toxicity, hypoxia, the very fast consumption of cellular energy associated to mitochondrial dysfunction activates anaerobic glycolysis which results in an intracellular pH decrease. The incoming acidosis is partially contrasted by the exchanges H+/Na+ and Na+/HCO3- with consequent influx of sodium. Because of the low ATP availability, sodium cannot be exchanged by the Na+/K+-ATPase and rapidly accumulates within the cell. The consequent osmotic load results in cell swelling which blocks the apoptotic processes known to require a reduction of the cell volume. This osmotic stress is worsened by the increase of cytosolic Ca2+ and ultimately results in the plasma membrane rupture.21
Finally, some mechanisms of drug-induced hepatic cell necrosis act by promoting nucleotides alterations or protein synthesis damage. In most cases, these events often follow drug-induced mitochondrial injury.22 However, selective nucleotide alterations have been described after Amanita Phalloides intoxication, as well as specific oxidation of protein sulphydril groups (y-glutamyl synthetase, glucose 6-phosphate dehydrogenase) are selectively provoked by some drugs; for example, damages to ATPase have been observed after cisplatin administration.23 Therefore, several antineo-plastic medicaments may cause hepatocellular necrosis through interference with the nucleotide metabolism.
The role of mitochondriaMitochondria, very complex intracellular organelles essential for eucariotic cell survival, frequently represent the main target for the toxicity of many substances.24,25 When severe imbalances of the mitochondrial energy metabolism or redox status occur, mechanisms leading to cell apoptosis or necrosis are consequently activated.26 In this context, the mitochondrial GSH pool plays a major role.12,27
Within mitochondria GSH acts by detoxifying free oxygen radicals physiologically generated by the respiratory chain. GSH also contributes to maintain in the reduced status the sulphydril groups of membrane proteins and enzymes, among which the ATP synthase complex and the Ca2+ dependent ATPase which pumps Ca2+ within mitochondria in order to keep low the cytosolic concentration. Cytosolic GSH depletion is not sufficient alone to determine a lethal damage to the cell. The latter event may occur when the total cellular content of GSH falls below 15%, thus involving the mitochondrial pool, which in fact represents per se approximately 15% of the total cellular GSH amount.28
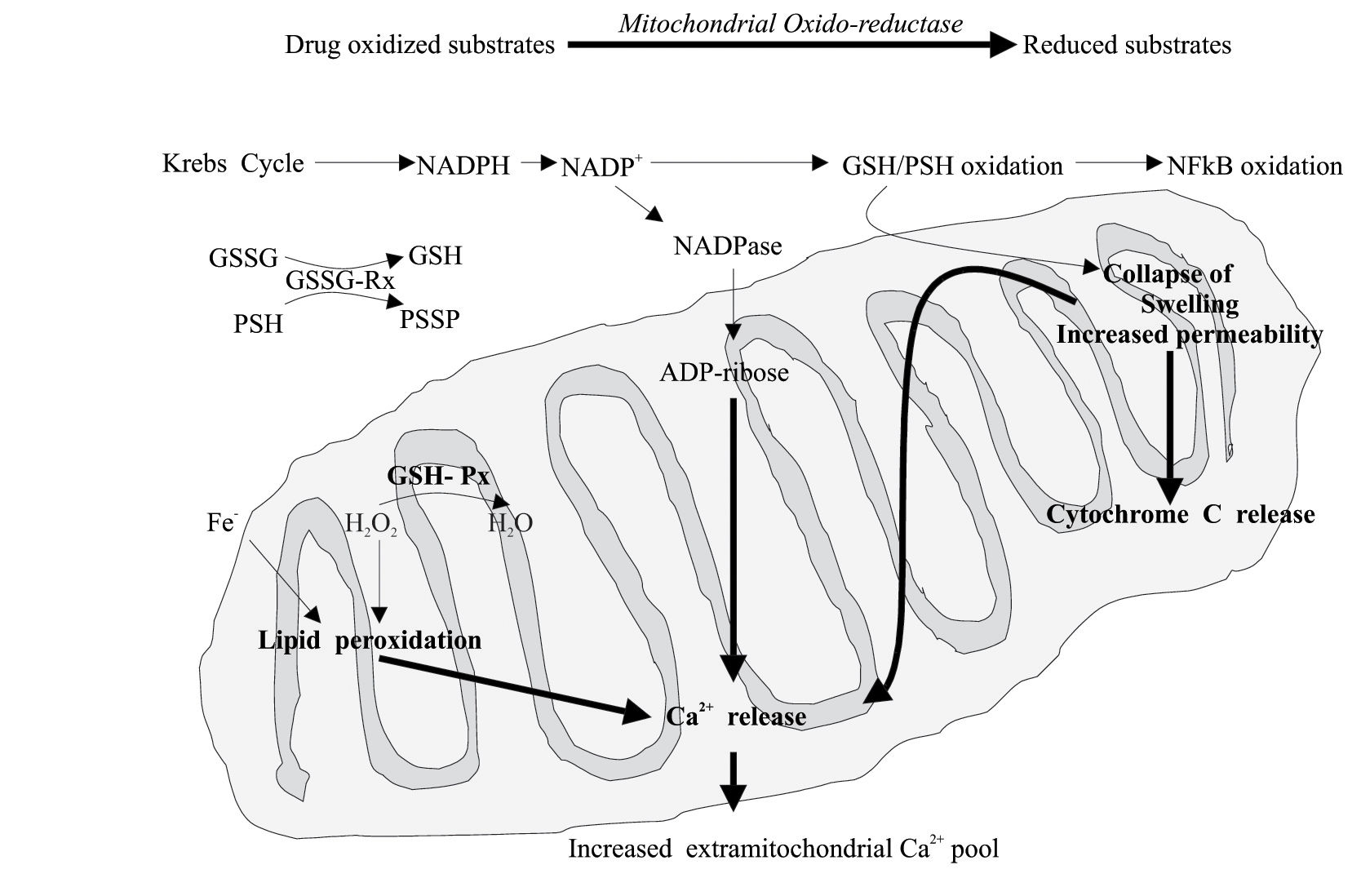
Recently, it has been suggested that the capacity of a drug to induce cell death by necrosis or apoptosis may depend on the extent and characteristics of the mitochondrial injury. Several mitochondrial factors have been associated to apoptosis or necrosis. It is supposed that apoptosis occurs mainly in sensitized cells such as those deficient in GSH.24 By contrast, the occurrence of the “mitochondrial permeability transition” (MPT) pore opening appears to be associated with apoptosis or necrosis according to the presence or deficiency of ATP.29 Another hypothesis suggests that it is the number of mitochondria developing MPT that activates the necrotic or the apoptotic death process in the presence of an uncoupling effect of the drug on the mitochondrial oxidative phosphorylation. However, it is generally believed that apoptotic cell death depends on the action of initiator and effector caspases; whereas necrotic cell death is associate with profound alterations of mitochondria and loss of ATP, while ATP is required for apoptosis. Therefore, the switch from apoptosis to necrosis may be determined by a sufficient mitochondrial impairment to deplete cell ATP and sufficient oxidative stress to inactivate caspases.30 Finally, the oxidation of mitochondrial protein and GSH induce oxidation of the NF-kB (an antiapoptotic factor), collapse of the transmembrane conductance, lipid peroxidation, mitochondrial swelling, cytochrome C and other pro-apoptotic molecule release, decreased uptake of Ca2+ and the consequent increase of cytosolic Ca2+ concentration22(Figure 4).

Drugs or toxic metabolites can cause mitochondrial alterations at different levels. The following impairment of the energetic and redox balance finally triggers apoptotic or necrotic processes. Mitochondrial permeability transition pore opening favors necrosis or apoptosis according to a poor or sufficient ATP level. GSH, reduced glutathione; GSSG, oxidized glutathione; GSH-Px, glutathione peroxidase; GSSG-RX, glutathione reductase; NFkB, nuclear factor kB; PSH, protein sulphydrils.
In Table III, the specific sites at which drugs may induce liver mitochondrial injury are reported.
Specific levels of drug-induced toxicity to liver mitochondria.
| • Inhibition of the electron transport chain: Amiodaron, Flutamide, Buprenorphin; |
| • Uncoupling of the oxidative phosphorylation: Amiodaron, Buprenorphin; |
| • Opening of the mitochondrial permeability transition pore: Valproate, Salicylates; |
| • DNA oxidation: Ethanol |
| • Inhibition of DNA synthesis: Zidovudin, Ribavirin, Fialuridine; |
| • Interference with the ß-oxidation: Valproate; |
| • Alteration of the transmembrane fatty acids transport: Ifosfamide. |
Not yet developed on large scale, but extensively used in human beings for experimental purposes, some breath test analyses using substances labeled with stable isotopes, are able to investigate selectively some specific mitochondrial functions. Accordingly, differences in the isotope exhalation may lead to the identification of specific alterations of mitochondrial metabolism induced by the drug.31
The role of hepatic non parenchymal hepatic cellsMore and more evidence suggest the participation of non parenchymal hepatic cells in the drug-induced hepatocellular injury.32 The involvement of non parenchymal hepatic cells depends on several factors among which the intrinsic characteristics of the damaging substance, the dosage, the production of reactive metabolites, the local oxygen tension.1,33
When high doses of a toxic compound reaches the liver, the injured hepatocytes can develop necrosis either because of intracellular alterations or as a consequence of the attack coming from free oxygen and nitrogen radicals released by activated Kupffer and endothelial cells.34 These cell populations can be both activated by chemotactic factors (i.e. leukotriene B4) released in turn by the same damaged hepatocytes35(Figure 3). Conversely, in the presence of prolonged exposure to moderate “subtoxic” doses of a hepatotoxic substance, the “stressed” hepatocytes (mitochondrial uncoupling, reduced content of GSH and ATP, lipid and protein oxidation, intracellular Ca2+ homeostasis imbalance) can undergo the attack of Kupffer cells and endothelial cells, which have been directly activated by the same toxic compound. (Figure 2) In this case both Kupffer and endothelial cells may release α-TNF and IL1B. In support to this hypothesis, it has been shown that the inhibition of macrophages activation or the use of α-TNF antagonists protect hepatocytes against acetaminophen and carbon tetrachloride hepatotoxicity, respectively.14,36
The role of the immune systemAcute idiosyncratic reactions can follow the ingestion of practically any drug compound and can result in hepatic necrosis. Both the exogenous molecule or its metabolites can activate an immune response in the host organism.22 Once reached the circulatory system, the substance is processed by the antigen presenting cells in the central lymphoid tissue directly or after the appearance of aptens or new antigens on the hepatocyte membrane. The latter case generally follows a covalent binding of the exogenous molecule with membrane constituents or even with intracellular proteins.1
Effector cells are B-lymphocytes, which release immunoglobulins and kinines and activate the complement cascade, and T-lymphocytes, which produce lymphokines (CD4) or determine direct cytotoxicity (CD8), resulting both in a condition of immunological mediated liver injury.1,22 Once the immune system has been activated, inflammatory mediators modulate the extent of the liver injury. In fact, it has been observed that the administration of prednisolon, a lymphocyte activation inhibitor, reduces the extent of drug induced liver necrosis.37 The local oxygen tension may have an important role in the progression of the immune-mediated toxic liver injury. For example, the metabolism of halotan in anaerobic conditions “reductive pathway’ may result in a mild hepatitis, whereas in the presence of high oxygen tension” oxidative pathway’ the exposure to halotan may be followed by a massive liver necrosis.33 The different behavior of this compound is explained with a higher immunogenicity of its oxidized metabolite which forms adducts with proteins.
Conclusions and future perspectivesAlthough the knowledge on mechanisms leading to drug-induced hepatic cell death is still incomplete, the occurrence of hepatic cell necrosis or apoptosis represents an event which follows the capacity of liver response to toxic insults. A growing number of observations indicates that the inflammatory reaction plays a key role in determining xenobiotic-induced liver damage. In any case, the research for a correct definition of the underlying mechanisms responsible for necrosis/apoptosis activation helps to clarify mechanisms of cell death. Such observations open to intriguing perspectives for treating toxic liver injury. Future issues might include the use of cytokine and death receptor antagonists, as well as strategies directed at upstream of mitochondria. Finally, approaches promoting liver regeneration and survival gene expression may overcome drug-induced cell death.



