



Primary hepatic amyloidosis (PHA) is characterized by abnormal deposition of monoclonal immunoglobulin light chains (AL) in the liver. This rare condition is frequently undiagnosed or misdiagnosed and can be associated with poor prognosis. At present, the precise pathogenesis is not fully understood. Despite that hepatomegaly and elevated alkaline phosphatase (ALP) are present in most patients with PHA, no specific clinical markers have been identified. Staining of hepatic tissues with Congo Red is often regarded as the “gold standard”. Pharmacological therapy should aim to rapidly reduce the supply of misfolded amyloidogenic AL. High-dose intravenous melphalan (HDM) and autologous stem cell transplantation (ASCT) appear to be the most appropriate therapy but controversies still exist.
Systemic amyloidosis is a group of disease characterized by deposition of various amyloid fibrils in the extracellular matrix and vessel walls in kidney (46%), heart (30%), liver (9%), spleen (5-10%), and gastrointestinal tract (7%).1-3 The two most common forms of systemic amyloidosis are primary and secondary amyloidosis. According to the new identification, systemic amyloidosis may be classified based on the deposition of immunoglobulin light chain (AL), serum amyloid A (AA), apolipoprotein A1 (Apo A1), and transthyretin (ATTR) type of fibrils. In primary amyloidosis, abnormal deposition of AL (к or X light chain) in the target organs is the key pathogenic mechanism. Thus, this type of amyloidosis is also termed AL amyloidosis. In contrast, secondary amyloidosis largely results from chronic inflammation, immunological disorders, and neoplastic diseases. Hepatic amyloidosis is the hepatic manifestation of systemic amyloidosis. In primary hepatic amyloidosis (PHA), also termed hepatic AL amyloidosis, there is an excess deposition of monoclonal immunoglobulin light chains or their fragments in the liver tissues, mostly in the hepatic perisinusoidal space (space of Disse), parenchyma, portal vessels, central vein, portal stroma and intercellular space. Long-term deposition of amyloid materials in these compartments leads to hepatomegaly, liver dysfunction, ascites, portal hypertension, liver failure, and even death.4,5
In the current report, we briefly reviewed the published data on PHA with the incorporation of the clinical, laboratory, and pathological features derived from patients with PHA encountered in our center.
EpidemiologyA precise estimation for the incidence rate of systematic amyloidosis can be difficult as many cases are frequently undiagnosed or misdiagnosed. In western countries, the reported incidence rate of syste-mic amyloidosis was 0.09-0.8% in hospitalized patients and 0.4-0.5% in autopsy cases.6 In China, systemic amyloidosis has been found in only 0.0140.03% of hospitalized patients.7 This relatively lower incidence in Chinese population may reflect the impact of multiple factors, such as genetic background, environment, life style, and insufficient awareness of the physicians. The incidence for primary amyloidosis is even lower, with an estimated (and perhaps an underestimated) age-adjusted incidence of 5.112.8 per million per year, and only 0.07%c in all hospitalized patients.8 Primary amyloidosis is two times more common in males than in females and usually affects the patients in their 6th to 7th decades, with a median age of diagnosis being 63-64 years.8 Although younger patients do get affected.9 Approxi-mately 9% of patients with amyloidosis have liver involvement,1 and more than 30% of patients with systemic amyloidosis have clinical evidence of hepatic involvement.10 Autopsy studies have reported an even higher incidence: 56-95% of patients with systemic amyloidosis exhibited hepatic involvement.2,10,11 Hepatomegaly has been reported in about 81-92% patients with hepatic amyloidosis,11 but this does not necessarily indicate the presence of PHA, especially when patients are complicated with congestive heart failure. In the past 10 years, we only encountered five patients with PHA confirmed by histopathology (4 males and 1 female), which approximately account for 15 patients per million in contemporaneous total hospitalized patients and 15.6% in the all amyloidosis patients of our hospital.
Classification and PathogenesisIn 2000, Falk, et al. systematically divided amyloidosis into five types: primary amyloidosis, se-condary amyloidosis, familial amyloidosis, amyloidosis of aging (or senile amyloidosis), and dialysis related amyloidosis.12 Up to now, at least 27 precursor proteins have been identified as causative agents of systemic amyloid diseases.13 Accordingly, systemic amyloidosis has been recently classified as AL, AA, Apo A1, ATTR, and other types. PHA refers to a constellation of symptoms and signs caused by the deposition of the AL amyloid materials in the liver.
Protein misfolding, aggregation, and fibril formation are important mechanisms for amyloidosis. Formation of precursor proteins, concentration in tissues, and toxicity to cells are the premise of amyloidosis. The interaction between amyloid proteins and local tissues/cells or extracellular matrixes is essential for disease development.14 In AL type of systemic amyloidosis, the amino acid sequence of the highly polymorphic light chain may be an important determinant for forming amyloidosis and organ se-lection.15,16 However, the precursor protein sources, heterogeneous proteins producing mechanisms, organ selectivity, and the exact mechanism of cellular and tissue damage have not been fully understood.
Pathological Features of PhaDeposition of amyloids in the liver may cause some characteristic pathological features. Macroscopically, the liver may look waxy gray with a smooth surface, blunt edge, moderate hardness, and poor elasticity. In advanced cases with massive amyloid infiltration, the liver may swell and become rubbery in consistency, giving a “lardaceous liver” appearance on cut surface. Splenomegaly is often present as a result of portal hypertension. Out of five patients in our hands, two had marked splenomegaly.
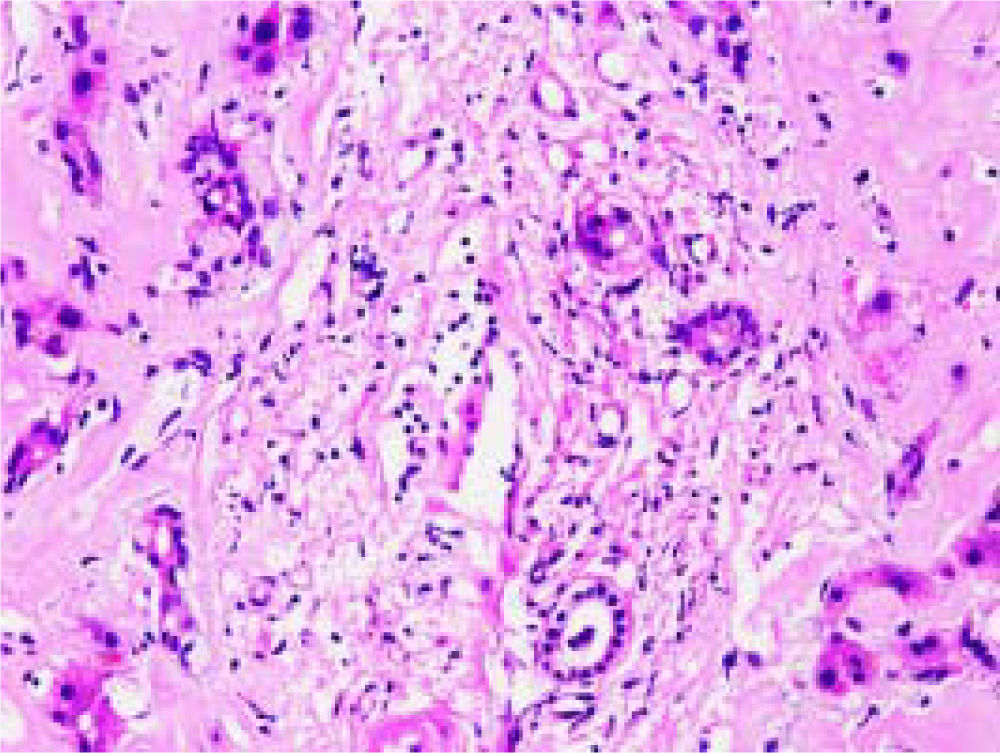
Microscopically, amyloid deposits are usually found in the hepatic parenchyma, along the sinusoid within the space of Disse, or in vessel walls. Hepatocytes are severely compressed by extensive accumulation of amyloid, leading to cellular atrophy and diminished cell number. A typical hematoxylin and eosin (H&E) staining of the liver tissue in a patient with PHA is shown in figure 1, in which heavy deposition of eosinophilic amyloid materials is visible in the extracellular matrix, with hepatic plates distortion and hepatocytes atrophy.

H&E stain of the hepatic tissue showing diffuse amyloid deposition. Eosinophilic amyloid materials are diffusely present in extracellular matrix, such as hepatic sinusoids, portal area, and vessel walls. Hepatic plates are distorted and hepatocytes atrophy. Original magnification: ×400.
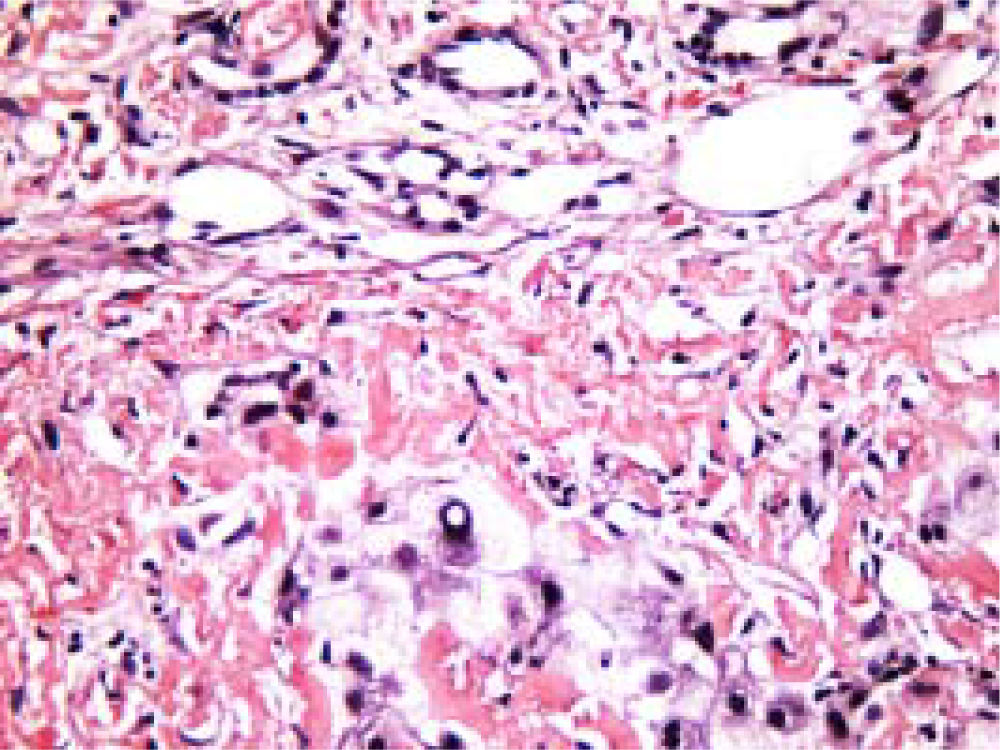
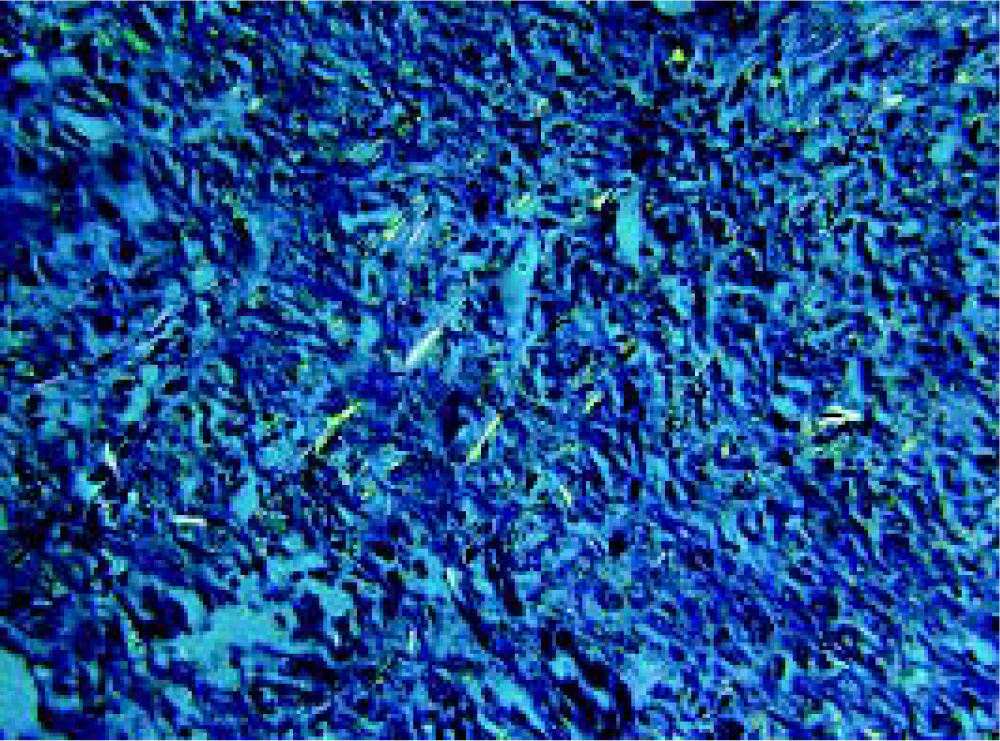
Staining of the hepatic tissues with Congo Red is often regarded as the gold standard for the definite diagnosis of hepatic amyloidosis. Under the light microscopy, Cong Red stained amyloid deposits may be diffusely present around hepatic plates and hepatic sinusoids (Figure 2). In PHA, amyloid deposition may be visible in liver parenchyma and vasculature. Typical AL amyloid materials may show as apple-green birefringence under the polarizing microscopy after the Congo Red-stain (Figure 3). The apple-green birefringence is usually resistant to potassium permanganate (KMnO4), a diagnostic feature of PHA.

Characteristic Congo Red staining of hepatic tissue showing amyloid protein deposit in the extracellular matrix. Amyloid protein spherical bodies can be found occasionally in hepatic sinusoids and intercellular space. Rudimentary and degenerated hepatocytes are visible. Original magnification: ×400

As amyloidosis can affect multiple organs, this disorder may present with a variety of clinical symptoms and signs, such as fatigue, weight loss, early satiety, nephrotic syndrome, edema, congestive heart failure, peripheral neuropathy, hepatomegaly, and hepatic failure.2,4,17 In PHA, although abdominal distension, fatigue, and weight loss are common presenting symptoms, and hepatomegaly, splenomegaly and ascites are frequently seen, these are all nonspecific. As a result, these manifestations are often mistakenly attributed to viral hepatitis, alcoholic liver disease, liver cirrhosis, or hepatic carcinoma. It is worth mentioning that although up to 81-92% of patients with systemic amyloidosis may have hepato-megaly,11 this does not necessarily indicates the presence of PHA, as liver involvement may be a part of the spectrum of secondary amyloidosis or result from congestive heart failure. Consistent with previous reports, in the five patients we encountered, the most common presenting symptoms and signs are abdominal distention (4/5), hepatomegaly (4/5), fatigue (3/5), anorexia (3/5), pitting edema of lower extremities (2/5), ascites (1/5), and oliguria (1/5). Two patients present with both splenomegaly and hepatomegaly. The degree of liver enlargement does not correlate with the severity of the disease, nor does it correlate with liver function abnormality and patient survival. PHA may be complicated by portal hypertension, varices and upper gastrointestinal hemorrhage, but these complications rarely worsen the already poor prognosis. In contrast, the presence of intrahepatic cholestasis and jaundice is rare but may signify a short survival.18
Laboratory testsElevation of alkaline phosphatase (ALP) is the most common laboratory finding, which is usually accompanied by elevated serum y-glutamyltransferase (GGT). Hyperlipidemia has also been described as the first biochemical manifestation of PHA. Hyperbilirubinemia and coagulation abnormalities may occur in patients with end-stage decompensated cirrhosis and hepatic failure. Other abnormalities such as thrombocytopenia, prolonged prothrombin time (PT), increased erythrocyte sedimentation rate, elevated alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST), reduced serum albumin and abnormal complement fragment have been reported, but all lack specificity for PHA. Ele-vation of monoclonal immunoglobulin in urine and blood as detected by protein electrophoresis and/or immunofixation electrophoresis may be very useful in the diagnosis of amyloidosis and in the follow-up of AL patients after chemotherapy,19 however not all patients may give positive results.9
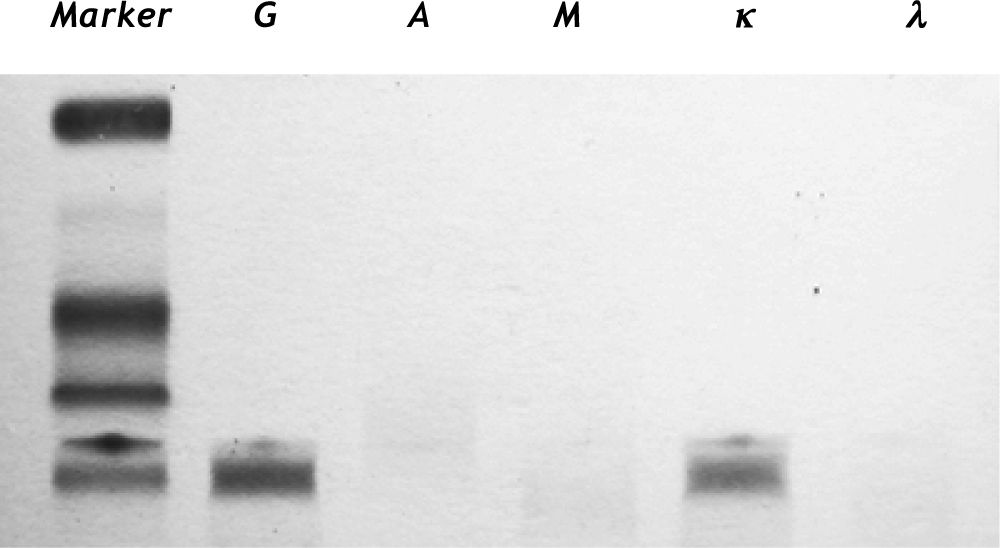
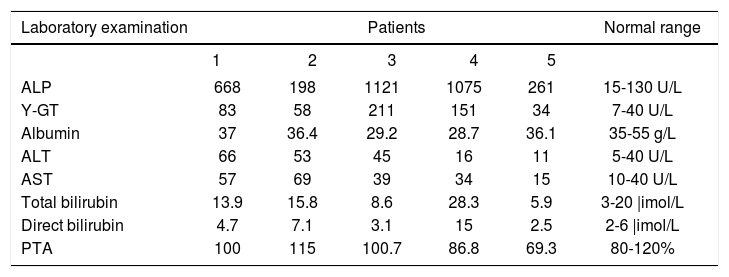
Typical laboratory findings are exemplified in table 1, in which common laboratory data from our patients are shown. Patients may exhibit increased serum and urine levels of y-globulin. By serum im-munofixation electrophoresis, the major component of immunoglobulin G (IgG) can be further identified. Increased к light chain of IgG is a typical finding (Figure 4). Other laboratory tests such as bleeding time, clotting time, cardiac enzymes, hemodiastase, urinary amylase, and anti-extractable nuclear antigens may all be within normal ranges at the early stage.
Typical laboratory data in five patients with hepatic amyloidosis
| Laboratory examination | Patients | Normal range | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| ALP | 668 | 198 | 1121 | 1075 | 261 | 15-130 U/L |
| Y-GT | 83 | 58 | 211 | 151 | 34 | 7-40 U/L |
| Albumin | 37 | 36.4 | 29.2 | 28.7 | 36.1 | 35-55 g/L |
| ALT | 66 | 53 | 45 | 16 | 11 | 5-40 U/L |
| AST | 57 | 69 | 39 | 34 | 15 | 10-40 U/L |
| Total bilirubin | 13.9 | 15.8 | 8.6 | 28.3 | 5.9 | 3-20 |imol/L |
| Direct bilirubin | 4.7 | 7.1 | 3.1 | 15 | 2.5 | 2-6 |imol/L |
| PTA | 100 | 115 | 100.7 | 86.8 | 69.3 | 80-120% |
ALP, alkaline phosphatase; g-GT, gamma-glutamyltransferase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; PTA, prothrombin time activity.
 and kappa light chain. The presence of an IgG kappa band in the gamma region was shown on serum immunofixation electrophoresis (IFE).")
Imaging studies constitute an integral part of diagnostic workup for amyloidosis. Due to a lack of specific imaging feature,20 PHA has frequently been misdiagnosed. The abdominal ultrasonography may show heterogeneous echogenicity of liver parenchyma. CT findings include diffuse or focally decreased parenchyma attenuation and triangular-shaped hepatomegaly. When coupled with clinical features, high resolution CT, dynamic CT and MRI may help differentiating amyloidosis from other infiltrative diseases based on the above features.20,21 Unfortunately, none of the present imaging techniques is capable of specifically demonstrating the presence of amyloidosis.20,22 Calcification may be seen, although rarely, in the hepatic parenchyma. Even though cholestasis can be detected in some patients, imaging studies usually show no dilatation of intrahepatic bile ducts. In systemic amyloidosis, increased accumulation of technetium-99m-labeled methylene diphosphonate (99mTc MDP) may be seen in affected organs. Thus, use of 99mTc MDP bone scintigraphy has been suggested as a screening test for the extent and the distribution of organ involvement in systemic amyloidosis. Recently, it was reported that high score of liver stiffness measured by Fibro Scan technique may be of value in patients with known amyloidosis, especially in whom liver biopsy is unavailable. However, it is difficult to distinguish amyloidosis from other cirrhotic liver disease.23
Diagnosis and differential diagnosisDiagnosis of amyloidosis can be difficult as patients with this disorder may present with symptoms that could closely mimic many other conditions. In addition, no specific laboratory test and imaging are available for this disorder.24 Thus, in the diagnosis of PHA, high index of suspicion is necessary. Generally, in any patients who present with hepatomegaly but without heart failure, with increased serum ALP but without abnormal imaging data, and with unexplained fatigue, edema, and weight loss, hepatic amyloidosis should be suspected. Currently, Congo Red staining of the amyloid fibrils is considered the gold standard for diagnosis of amyloidosis. Once the histological diagnosis of hepatic amyloidosis is established, the amyloid type should be defined for unequivocal identification of the deposited amyloidogenic protein in order to avoid misdiagnosis and inappropriate treatment.
In the definite diagnosis of PHA, differential diagnosis should always be considered comprehensively. Other common liver diseases such as viral hepatitis, alcoholic hepatitis, and Wilson's disease should all be on the differential list. Relevant investigations such as serological examinations for hepatitis B virus surface antigen, hepatitis C virus antibody, human immunodeficiency virus antibody, cytomegalovirus antibody, and Epstein-Barr virus DNA should be tested to exclude common viral hepatitis before hepatic amyloidosis is confirmed. Furthermore, fatty liver disease, autoimmune liver disease, drug induced liver disease and hepatocellular carcinoma should all be excluded.
TreatmentEarly diagnosis is a key to effective therapy as it allows reversal of the organ damage and better tolerability of chemotherapy.25 Unfortunately, no uniformly effective and specific treatment for PHA is available so far.26 The relatively effective approach can be achieved by chemotherapy, as described below. Meanwhile supportive measures may minimize treatment toxicity and support the organ functions.
The combination therapy with low-dose oral melphalan plus prednisone (MP) has previously been re-garded as a standard therapy for AL. But the time to response is more than 12 months in about 30% of patients. For these patients, oral melphalan plus dexamethasone (MDex) may induce a rapid response in 67% of AL patients ineligible for autologous stem cell transplantation (ASCT) due to advanced disease. The MDex regimen is a safe modality, with only 4% treatment-related mortality.27 Apart from the MDex approach, high-dose intravenous melphalan and ASCT (HDM/ASCT) has been suggested as the first-line treatment for selected patients with AL amyloidosis nowadays.10 In addition, treatment with HDM/ASCT did not increase treatment-related mortality or reduce the hematologic response rate.10 Thus, HDM/ASCT may be considered an appropriate treatment for PHA, although controversy does exist.28
Liver transplantation has also been successfully performed in patients with end-stage of hepatic amyloidosis, but it alone does not eliminate the source of amyloid production, which means an inevitable recurrence will occur sooner or later. Other new treatment modalities, such as small interfering RNA and gene conversion strategies, have proved successful in reducing the synthesis of amyloidogenic light chains in experiments.29 More efficient therapy for hepatic amyloidosis is still a subject of extensive clinical research.5
PrognosisPrognosis is poor in patients with hepatic amyloidosis, with a median survival of 10-14 months from diagnosis.30 However, untreated PHA has a mean life span of 8-9 months and the projected 5-and 10-year survival rate is only 13-17% and 1-7% respectively.11 Some patients may benefit from systemic chemotherapy but multiorgan failure is a contraindication. Changes in serum-free light chain might better predict outcome compared to changes in immunoglobulin levels in primary amyloidosis.31 According to the consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, a reduction in liver size by at least 2 cm as documented by radiographic or radionuclide imaging and 50% decrease in abnormal ALP value are the primary measures of hepatic response. In contrast, progression is defined as 50% increase of ALP above the lowest recorded value.32 The median survival time of our three patients who successfully completed melphalan and prednisone treatment (one combined with ASCT) was 3.7 years (1.5 to 5.2 years). These patients usually die of decompensated cirrhosis, hepatic failure and secondary infections.
ConclusionClinicians should be aware of this rare but potentially fatal disease when patients present with hepatomegaly, and/or increased serum level of ALP that can not be explained by common liver diseases such as viral and alcoholic hepatitis. Other organs and tissues such as heart, kidney, and bone marrow should also be investigated to clarify if systemic amyloidosis is present. Definite diagnosis of PHA is based on hepatic histopathologic examination following Congo Red stain. Once the amyloidosis is confirmed to be of light chain origin, ASCT or trials of appropriate systemic chemotherapy should be arranged promptly.25 Although no specific treatment to PHA is available at the present time, both quality of life and survival of the patients with amyloidosis have improved during the past three decades, likely as a cumulative result of earlier diagnosis, better risk assessment, and improved therapies and supportive care.8,30 However, the precise pathogenesis, effective treatment and availability of novel agents for decreasing high risk of early death should be the focus of future studies.
AcknowledgementsWe thank Professor Tai-Ling Wang from Department of Pathology of China-Japan Friendship Hospital, Beijing, China for her technical assistance in pathological diagnosis of liver disease.
Abbreviations- •
AA: serum amyloid A.
- •
AL: amyloid light chain.
- •
ALP: alkaline phosphatase.
- •
ALT: alanine aminotransferase.
- •
ASCT: autologous stem cell transplantation.
- •
AST: aspartate aminotransferase.
- •
CT: computerized tomography.
- •
GGT: Y-glutamyltransferase.
- •
HDM: high-dose intravenous melphalan.
- •
IgG: immunoglobulin G.
- •
MDex: oral melphalan plus dexamethasone.
- •
MP: melphalan plus prednisone.
- •
MRI: magnetic resonance imaging.
- •
PTA: prothrombin time activity.
- •
99mTc MDP: technetium-99m-labeled methylene diphosphonate



