






Primary hepatic neuroendocrine tumours are rare tumours effecting relatively young patients. As metastatic neuroendocrine tumours to the liver are much more common, extensive investigations are crucial to exclude a primary tumour elsewhere. We report a case of a 27 year old woman who presented with fatigue, increased abdominal girth and feeling of early satiety and bloating. Extensive work up failed to show tumour at another primary site. Hepatic artery embolization showed no effect, so the patient underwent total hepatectomy and live-donor liver transplant. Grossly the tumour measured 27 cm. Microscopic examination showed bland, monomorphic cells growing in tubuloglandular and trabecular growth patterns. Cells were positive for neuroendocrine (synaptophysin, chromogranin, CD56) and epithelial markers (MOC31, CK7, CK19). Cytoplasmic dense neurosecretory vesicles were seen on ultrastructural examination. Based on the Ki-67 rate, mitotic count, lack of marked nuclear atypia and absence of necrosis, a diagnosis of primary neuroendocrine grade 2 was conferred.
Neuroendocrine tumours have an annual incidence of 1 to 2 per 100,000, and represent 2% of all tumours in the gastrointestinal tract. The most frequent site of neuroendocrine tumours is in the gastrointestinal tract (followed by the bronchopulmonary system), accounting for approximately 70% of the total neuroendocrine tumours in the body. The most common sites are the appendix, ileum and rectum, followed by the colon, stomach, duodenum and jejunum.1-3 Neuroendocrine tumours frequently metastasize to the liver, but the liver itself seldom is the site of a primary tumour. A review of the English literature on neuroendocrine tumours in 2005 showed only 95 cases of primary hepatic neuroendocrine tumours.3 Since then, 5 other cases have been reported.4-8
Clinicopathological findings as summarized by previous reviews1-4 articles include:
- •
Patients are relatively young (average 45 years of age, range of 8-83), with no gender predominance.
- •
Clinical presentation varies considerably ranging from no symptomatology, to symptoms secondary to mass effect of the tumour on the hepatic parenchyma and adjacent structures (abdominal pain, weight loss, palpable mass, gastric outlet obstruction). The pathognomonic features of carcinoid syndrome which occur from metastatic neuroendocrine tumours to the liver, occur infrequently (despite evidence showing the presence of bioactive amines) with primary hepatic neuroendocrine tumours (5-10%).
- •
Tumours arise in a background of non-cirrhotic liver and range in diameter from a few centimeters to the largest previously reported as being 26 cm.9
- •
Pathologic diagnosis requires routine hematoxylin and eosin (for general morphology and grading) with ancillary studies demonstrating evidence of neuroendocrine differentiation.
- •
A variety of therapeutic approaches have been used (hepatic lobectomy, systemic chemotherapy, hepatic artery chemoembolization, octreotide alone or with surgery), with surgical removal regarded as the most effective.
- •
5 year survival rates are approximately 10-20%, but more favorable outcomes have been documented in aggressive treatment with liver resection and orthotopic liver transplant.10
A healthy 27 year old woman presented with fatigue, increased abdominal girth and feeling of early satiety and bloating in April 2010. On examination her right hemidiaphragm was raised with decreased air entry to the right base and right middle lobe, the liver edge was palpable at the level of the umbilicus in the mid-clavicular line. There were no stigmata of chronic liver disease. Clinical examination and an extensive work up (which included PET scan) failed to show tumour at another primary site. A CT scan performed in June 2010 (Figure 1) showed multiple cystic lesions with the overall mass measuring 24 x 14 cm which was involving primarily the caudate and right lobe of liver with sparing of segment 2/4, parts of segments 4, 5 and 6. She was scheduled for drainage of these cysts as the etiology was believed to be Echinococcus infection. Intraoperatively however, extensive tumour mass was seen and a biopsy was performed, with a subsequent diagnosis of gland forming intrahepatic cholangiocarcinoma.
 × 15.7 cm (AP) mass involving most of the right liver lobe with extension in to the medial segment of the left liver lobe.")
Pertinent investigators include negative Echinococcus and hepatitis serology, normal serum levels of chromogranin A, alpha-fetoprotein, and carcinoembryonic antigen. CA19-9B was elevated to 69 kU/L (normal < 37 kU/L). A repeat CT scan in October 2010 and MRI performed in November 2010 demonstrated no interval change in the large heterogenous liver mass, the radiographic impression was that the enhancement characteristics and the appearances were highly suggestive of a hepatocellular carcinoma. Cholangiocarcinoma was considered less likely due to the relative lack of hepatic duct dilatation and capsular retraction. The patient underwent a hepatic artery embolization procedure in December 2010; however, a follow up post embolization CT scan performed in January of 2011 demonstrated no effect.
She underwent total hepatectomy and live-donor liver transplant. Immunosuppression consisted of tapering corticosteroids, mycophenolate mofetil and tacrolimus. Intraoperatively, frozen sections performed on multiple enlarged nodes and diaphragmatic nodules failed to show tumour implants or extrahepatic disease. She was transferred to the ICU post-operatively and did well. A day after a re-look laparotomy (performed on post-operative day 2), she was extubated and transferred to the ward. Post-operatively she had minor complications but with quick resolution. On follow up 6 months post-transplant, she was doing well with no evidence primary tumour recurrence or metastases.
Gross examination (Figure 2) of the massively enlarged explanted liver (5,470 g) showed that the entire right lobe and one half of the left lobe replaced by a large multinodular tumour measuring 27 cm (transverse) x 22 cm (anterior to posterior) x 12 cm (superior to inferior). The nodules were white, yellow with focal hemorrhages, central scarring and were separated by thin fibrous septae. A large 8 cm cyst was seen in the centre of the lesion. The majority of the tumour was grossly viable. There was no rupture of the tumour capsule and there was no gross invasion into the porta hepatis, region of the bile duct and portal vein, and hepatic vein.
.")
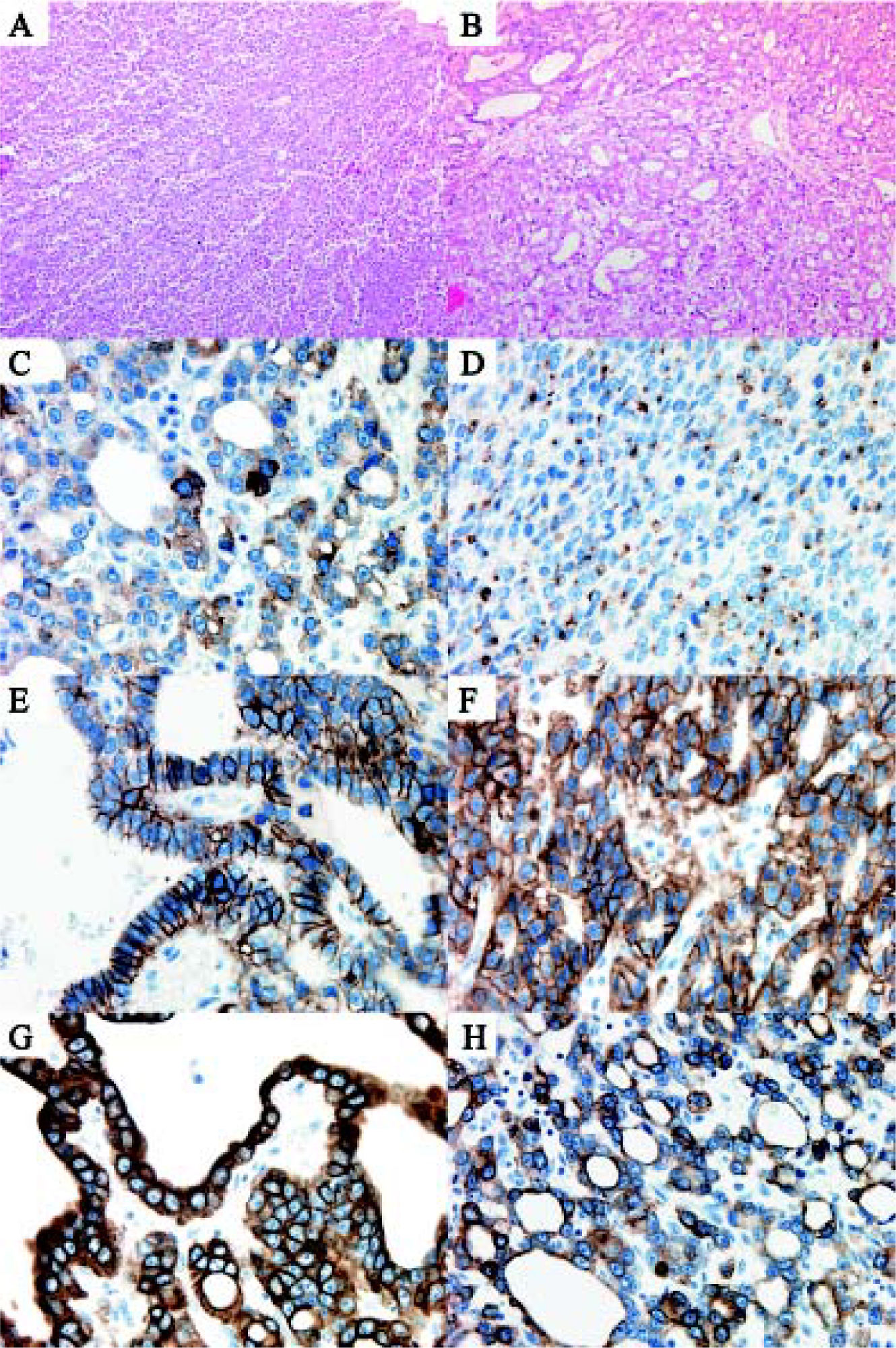
Microscopic examination showed a hepatic neoplasm with an unusual growth pattern with two separate histological patterns that blended into one another arising in a background of non-cirrhotic liver. The first pattern had well-developed tubuloglandular architecture with blood and eosinophilic secretions filling tubular spaces (Figures 3A and 3B). Tubules were lined by neoplastic cuboidal cells with large vesicular nuclei with prominent nucleoli and a moderate amount of eosinophilic granular cytoplasm. This component of the tumour had scanty mitoses (maximum of 3 per 10 high power fields) and the tubuloglandular structures were arranged in a back-to-back pattern with minimal or no intervening stroma or desmoplastic reaction. The second pattern observed was a solid sheet-like pattern punctuated by delicate fibrovascular cores which imparted focally a papillary appearance. In this solid portion of the tumour, there were some areas with a trabecular architecture. The sheets of cells in the solid component had identical morphology to the cells in the tubuloglandular area. There was no tumour necrosis and no vascular invasion. Scanty possible mucin in the lumina of some tubules as well as intracytoplasmic globules was demonstrated by PAS diastase stain. The mucicarmine stain and immunohistochemical stains for mucin (MUC-1, MUC-2, MUC-5AC) however failed to show positivity for mucin. In both components, neuroendocrine markers were positive (synaptophysin strongly positive, chromogranin focally positive, CD56 weakly focally positive) as was MOC-31 (in membrane fashion), CK7, CK-19, but the tumour was negative for CEA, Hepar-1, CK20, TTF-1, thyroglobulin and CD10 (Figures 3C-3H). The Ki-67 rate on average was 15% (focally up to 25%).
 and tubuloglandular architecture (B). Immunohistochemistry showing positive staining for: synaptophysin (C), chromogranin (D), CD56 (E), MOC-31 (F), CK-7 (G) and CK-19 (H).")
Electron microscopy was performed and in both the solid sheet-like as well as tubuloglandular portions of tumour, electron dense neurosecretory vesicles were demonstrated (Figure 4). Intriguingly, in both areas, the neoplastic cells showed similar ultrastructural features of microvilli formation and cells were attached to one another through tight junctions in their cell membranes.
 ranging in size from 220-350 nm were seen within the cytoplasm of tumour cells in areas of both trabecular (A) and tubuloglandular architecture (B). Cells lining the tubuloglandular components of the tumour (C) showed luminal microvilli (white arrow) and tight junctions (black arrow).")
Electron microscopy. Electron dense neurosecretory granules (black arrows) ranging in size from 220-350 nm were seen within the cytoplasm of tumour cells in areas of both trabecular (A) and tubuloglandular architecture (B). Cells lining the tubuloglandular components of the tumour (C) showed luminal microvilli (white arrow) and tight junctions (black arrow).
Extensive workup to exclude a primary tumour elsewhere was negative. Given the lack of marked nuclear atypia and necrosis, and an average of 3 mitosis per 10 high power fields and 15% Ki-67 index, the patient was diagnosed with a primary hepatic neuroendocrine tumour, grade 2 (see later for grading criteria).
DiscussionPrimary neuroendocrine tumours of the liver are extremely rare. The diagnosis of this tumour poses a challenge to clinicians, radiologists, and pathologists. Not only is it extremely rare with non-specific clinical and radiographic presentation, but it also is a heterogeneous entity histologically. In this patient, the radiographic diagnosis of Echinococcus infection followed by hepatocellular carcinoma, and the diagnosis of cholangiocarcinoma based on initial biopsies highlights this diagnostic challenge.
Neuroendocrine tumours originate from neuroendocrine cells from the embryological neural crest. In the gastrointestinal system, neuroendocrine cells are found from mouth to anus, including pancreas, but few in the biliary tree and none in the liver. The etiology and histogenesis of hepatic neuroendocrine tumours have remained elusive and controversial. Proposed theories on the origin of neuroendocrine cells that give rise to primary hepatic neuroendocrine tumours include ectopic neuroendocrine cells of pancreatic or adrenal origin, neuroendocrine cells from within the intrahepatic biliary tree, or from neuroendocrine-programmed ectoblasts.11,12
Microscopically neuroendocrine tumours show polygonal, monomorphic cells with finely granular nuclear chromatic with small or not visible nucleoli, and bland cytological features. However, some tumours can show relative pleomorphism. The type of growth pattern that is seen (A-insular/nested, B-trabecular, C-glandular/acinar, D-poorly differentiated) may be characteristic for neuroendocrine tumours for specific sites. In our case the tumour showed trabecular as well as glandular/acinar patterns, suggesting that the neoplastic neuroendocrine cells are either of hindgut or foregut derivation.
Given the lack of universally accepted nomenclature of neuroendocrine tumours of the gastrointestinal tract, a new classification scheme was introduced by the WHO in 2010. The term carcinoid in the 1980 WHO, is now divided into 3 separate tiers: neuroendocrine tumour grade 1 (NET G1), NET G2 and neuroendocrine carcinoma (NEC). A NET is a well-differentiated neoplasm that is separated into G1 (< 2 mitosis per 10 high power fields, and/or < 2% Ki-67 index) or G2 (2-20 mitosis per 10 high power fields, and/or 3-20% Ki-67 index) and encompasses neoplasms previously termed 'carcinoid tumour'. The high grade (G3) neuroendocrine neoplasm which shows greater nuclear atypia, multifocal necrosis and greater mitoses (> 20 mitosis per 10 high power fields, and/or > 20% Ki-67 index) is now termed 'neuroendocrine carcinoma' and refers to neoplasms previously classified as small cell carcinoma, large cell neuroendocrine carcinoma, and poorly differentiated neuroendocrine carcinoma.13 In our case, neoplastic cells showed mitotic count and Ki-67 index, consistent with a grade 2 NET.
The immunoprofile in this case was consistent with cells of a neuroendocrine origin (positive synaptophysin, chromogranin and CD56); however, expression of markers of epithelial differentiation (MOC31, CK7 and CK19) were also observed. Given the rarity of primary hepatic neuroendocrine tumours, there is no literature available regarding expression of epithelial markers in these tumours. However, expression of CK7 and CK19 in conjunction with neuroendocrine cell markers has been previously reported in extrahepatic neuroendocrine gastrointestinal tumours as well as neuroendocrine lung tumours. CK7 expression for example, has been shown in approximately 10% of GI (stomach and large intestine), 50% of pancreatic carcinoid tumours,14 and in the lung 5% of carcinoid tumours, 40% of small cell carcinomas and 70% of large cell neuroendocrine carcinomas.15 CK19 expression has also been shown in 80% of carcinoids from the appendix16 and in the lung 70% of carcinoid tumours and 80% of both small cell carcinomas and large cell neuroendocrine carcinomas show CK19 expression.15
Additional evidence of neuroendocrine differentiation was provided through electron microscopic examination. This method has been used in the past by other investigators to show uniform electron dense neurosecretory granules in the cytoplasm of neoplastic cells.17-19 In our case, we also observed these granules and also discovered that cells lining glandular cystic spaces had microvilli projecting in to lumens and these cells were attached to each other with tight junctions, a phenomenon which has been previously described.17
An important differential is cholangiocarcinoma, an entity which would require a much different chemotherapeutic approach should the lesion grow or metastasize. Typically, cholangiocarcinoma develops in a non-cirrhotic liver. Macroscopically it is classified into three types (mass-forming, periductal infiltration, and intraductal-growth type). Histologically cholangiocarcinoma are adenocarcinomas with tubular growth being the most common pattern. Micropapillary and acinar (or cord-like) growth patterns also occur. Tumour cells, which may be pleomorphic, are usually small to medium sized, cuboidal or columnar, with small nuclei and nucleoli and pale eosinophilic cytoplasm. An important characteristic is the presence of fibrous stroma with the central portion of the tumour being sclerotic and hyalinized. Histochemical stains such as mucicarmine, PASD, Alcian blue, and immunohistochemical stains for mucin core (MUC) proteins are positive. Tumour cells are also immunoreactive for cytokeratins, such as CK7 and CK19.
In summary, primary hepatic neuroendocrine tumours are rare tumours that effect relatively young patients and typically do not present with the carcinoid syndrome. They typically arise in a background of non-cirrhotic liver with diagnosis requiring routine hematoxylin and eosin as well as ancillary studies (primarily immunohistochemistry) demonstrating evidence of neuroendocrine differentiation. The exact etiology remains elusive; our knowledge on the range of morphology is still gaining experience and warrants further investigation.



