




Epigenetic changes represent a mechanism connecting external stresses with long-term modifications of gene expression programs. In solid organ transplantation, ischemia-reperfusion injury (IRI) appears to induce epigenomic changes in the graft, although the currently available data are extremely limited. The present study aimed to characterize variations in DNA methylation and their effects on the transcriptome in liver transplantation from brain-dead donors.
Patients and Methods12 liver grafts were evaluated through serial biopsies at different timings in the procurement-transplantation process: T0 (warm procurement, in donor), T1 (bench surgery), and T2 (after reperfusion, in recipient). DNA methylation (DNAm) and transcriptome profiles of biopsies were analyzed using microarrays and RNAseq.
ResultsSignificant variations in DNAm were identified, particularly between T2 and T0. Functional enrichment of the best 1000 ranked differentially methylated promoters demonstrated that 387 hypermethylated and 613 hypomethylated promoters were involved in spliceosomal assembly and response to biotic stimuli, and inflammatory immune responses, respectively. At the transcriptome level, T2 vs. T0 showed an upregulation of 337 and downregulation of 61 genes, collectively involved in TNF-α, NFKB, and interleukin signaling. Cell enrichment analysis individuates macrophages, monocytes, and neutrophils as the most significant tissue-cell type in the response.
ConclusionsIn the process of liver graft procurement-transplantation, IRI induces significant epigenetic changes that primarily act on the signaling pathways of inflammatory responses dependent on TNF-α, NFKB, and interleukins. Our DNAm datasets are the early IRI methylome literature and will serve as a launch point for studying the impact of epigenetic modification in IRI.
Solid organ transplantation (SOT) is the most effective therapy for end-stage organ disease. Crucial advances in organ preservation, immunosuppression therapies, anesthesiology management, and surgical techniques have contributed to the success of its implementation [1,2]. Dynamic cumulative injury of the graft is primarily associated with donor type, age, comorbidities, and maintenance, but ischemia-reperfusion injury (IRI) could worsen its development. Sustained injuries, surgical complications, recipient comorbidities, and host vs. graft immune response could collectively affect the SOT outcome [1]. Around 10% of early organ failures are attributed to IRI during liver transplantation (LT), leading to primary graft dysfunction and a higher incidence of acute and chronic rejection [3].
IRI triggers metabolic alterations, including reduction of cellular adenosine triphosphate levels, increase of intracellular/mitochondrial calcium levels, endoplasmic reticulum stress, and changes in pH homeostasis, etc. [4]. In addition, xenobiotics enter the liver after reperfusion, which activates Kupffer cells (KC). KC secrete proinflammatory cytokines, tumor necrosis factor-α (TNF-α), and interleukin-1β, triggering inflammatory cascades, cell apoptosis, and immune response activation [5].
In the advent of the omics era, several researchers approached the study of IRI using high-throughput technologies by transcriptomics [6], proteomics [7], miRNomics [8], metabolomics [9], and recently single-cell transcriptome analysis (scRNAseq) [10]. However, there are no studies on epigenetic modifications at the whole-genome level during IRI conditions in LT.
Epigenetic modifications have gained strong interest as a novel biomarker in transplantation [2]. The three main epigenetic modifications, namely histone modification, DNA methylation, and nucleosome positioning, are reversible changes to the genome (no alteration in the DNA sequence) [11].
To the best of our knowledge, this study is the first research on DNA methylation profiles aimed to characterize the effect of IRI in the epigenetic context during human liver transplantation. Gene expression profiling by RNAseq in the same biopsies was also performed and compared against the former in terms of functional enrichment profiles to identify differentially regulated biological processes / molecular pathways during IRI.
2Methods2.1Patients' enrollment and sample collectionTwelve whole liver allografts were procured from brain-deceased donors and transplanted in adult patients with end-stage liver disease from February 2022 - August 2022. Exclusion criteria comprised donation after circulatory death, split liver allografts; allografts managed with machine perfusion, LT for hepatocellular carcinoma, ABO incompatibility, and re-transplantation cases. Liver procurement was performed with a standard technique after systemic heparinization, aortic cannulation, and arterial cold flushing with Celsior preservation solution. Back-table surgery was performed, keeping the graft in an ice bath at 4°C. LT was performed using a caval-sparing approach. Graft reperfusion was antegrade and sequential, with a portal-first system. Cold ischemia time (CIT) is the interval between cross-clamp at procurement and graft placement into the recipient's abdomen; warm ischemia time (WIT) is the interval between graft placement into the recipient's abdomen and graft reperfusion. Immunosuppression regimen was based on tacrolimus and steroids. Tru-cut (16-gauge) biopsies of all liver allografts were serially collected at three-time points: during the warm phase of organ procurement (T0), during back-table surgery (cold ischemia, T1) while the graft was maintained in ice at 4°C deprived of blood; at transplant after graft reperfusion with recipient's blood, immediately before abdominal closure (T2). Liver samples were collected in RNA later solution and stored at -20°C for omics analysis. At T1, an additional biopsy was taken, and formalin was stored for histopathology examination (T1-F).
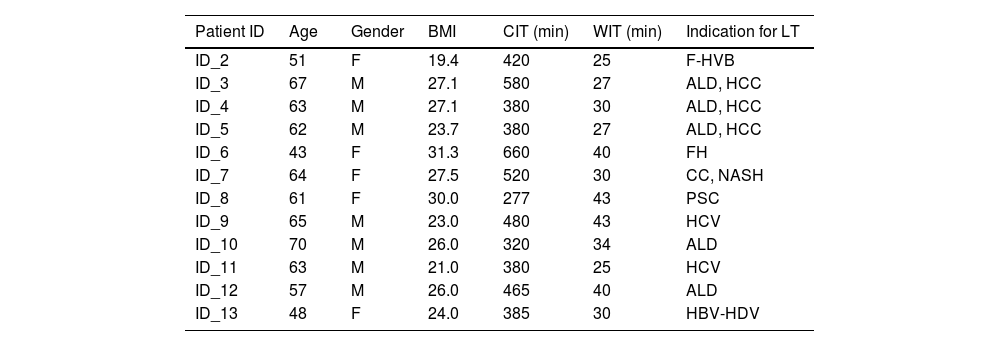
2.2Recipient and donor demographicsThe median recipient age at LT was 59 years (range 43-70), with a male-to-female ratio of 7:5 (Table 1). The most frequent causes of liver cirrhosis were alcohol abuse and viral hepatitis, with a median MELD score of 14 [8-40]. Liver grafts were procured from donors with a median age of 57 (32-71) and a male-to-female ratio of 5:7. The median CIT and WIT were 437 min (range 277 – 660) and 33 min (range 25 – 43), respectively while the median LT operative time was 370 [290-538]. Intraoperatively, a median of 2 [1-4] units of packed blood cells were transfused. No cases of primary non-function, early (within 90 days after LT) vascular or biliary complications were noted. Early allograft dysfunction, defined according to the MEAF score, was 3.3 [0.9-5.3]. Three patients developed biopsy-proven acute cellular rejection, successfully treated with steroid boluses. At a median follow-up of 16 months (13-17), 11 patients were alive with a preserved graft function. One patient developed graft biliary failure due to late arterial thrombosis and was successfully re-transplanted. Additional donor demographics and the description of the histological analysis of biopsies at T1 are reported in the supplementary information (Supplementary material (SM), Table S1).
Demographic, times of cold and warm ischemia, and indications for transplantation of each recipient patient
CIT, cold ischemia time; WIT, warm ischemia time, F-HVB, fulminant Hepatitis B Virus-related; ALD, alcoholic liver disease; FH, fulminant hepatitis; CC, cryptogenic cirrhosis; NASH, non-alcoholic steatohepatitis; PSC, primary sclerosing cholangitis; HCC, Hepatocellular carcinoma.
Thirty-six biopsies (12 at each time point) were subjected to DNA methylation (DNAm) and RNAseq analysis. Detailed methodological procedures from sample preparation to data analysis of the generated datasets are described in SM (section Methods). Briefly, DNA methylation profiles were computed based on single CpG measurement values and methylation levels in predefined regions such as gene promoters or CpG islands. All CpG positions were mapped against the human hg38 reference genome. For gene expression profiles by RNAseq, high quality sequences were aligned to the human reference genome hg38 and gene counts were extracted according to Gene annotations [12]. Raw DNAm and raw transcriptome datasets has been deposited into GEO and Sequence Read Archive accession number PRJNA1015016.
2.4Functional enrichment of methylation/transcriptome data, cell enrichment analysis, and validation of differential gene expression by qPCRGene Ontology (GO), Reactome (REACT), and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis were performed using the g: Profiler platform to visualize functional profiles of key gene sets [13]. GO terms for Molecular Function (GO:MF), biological process (GO:BP), and cellular component (GO:CC), as well as REACT and KEGG terms with P < 0.05, using Fisher's one-tailed test, were considered statistically significant and reported in the plots. Cell type (TC) specific enrichment was performed using WebCSEA, a one-click application that provides a comprehensive exploration of TC-specificity of genes among human major TC map [14]. See further information about TC enrichment analysis in SM (section Methods). Gene expression validation of the most up-and down-regulated genes, as well as for the most interesting candidates individuated in the study, were validated by qPCR. Detailed information about candidates’ selection and qPCR methodology for validation is described in SM (Supplementary Methods).
2.5Statistical analysesPairwise comparisons were performed between conditions (T1 vs. T0, T2 vs. T1, and T2 vs. T0), obtaining the differential methylation and gene expression profiles.
Site level-differential methylation was computed based on various metrics. For each site: a) the difference in mean methylation levels (mean. diff of β values) of the two groups being compared, b) the quotient in mean methylation, and c) a statistical test (limma) assessing whether the methylation values in the two groups originate from distinct distributions. Region level-differential methylation was computed based on the following quantities: the mean difference in means across all sites in a region of the two groups being compared (mean.mean.diff) and the mean of quotients in mean methylation as well as a combined p-value calculated from all site p-values in the region. Each site and region was assigned a rank based on the three quantified respective criteria. A combined rank was computed as the maximum (i.e., worst) rank among the three ranked criteria where lower-numbered ranks for a site or region have higher confidence of exhibiting differential methylation. Scatterplots of the site/promoter region group mean and volcano plots of each pairwise comparison, colored according to a given site's adjusted p-value (<0.05) and combined rank for each region, were obtained with RnBeads package version 2.12.2 [15,16]. The top 1000 promoter regions with the best-combined ranks were selected as differentially hyper/hypomethylated for downstream analyses [17].
Differential gene expression analysis was performed using default parameters of the DESeq2 (v.1.36) Bioconductor package [18]. To detect outlier data after normalization, R packages were used. Prior to differential gene expression analysis, we dropped all genes with low normalized mean counts to improve testing power while maintaining type I error rates. Estimated false discovery rate (FDR) values for each gene were adjusted using the Benjamini-Hochberg method. Protein-coding genes were considered significantly different expressed at FDR-corrected q < 0.05 and log2FC of ≥ |1|.
2.6Ethical statementAll biopsies were obtained as per protocol biopsies in the donor (T0 and T1) and in the recipients (T2) as post-reperfusion biopsy according to Center's specific protocol. All research was conducted following both the Declarations of Helsinki and Istanbul. Informed consent was obtained by all patients before transplantation.
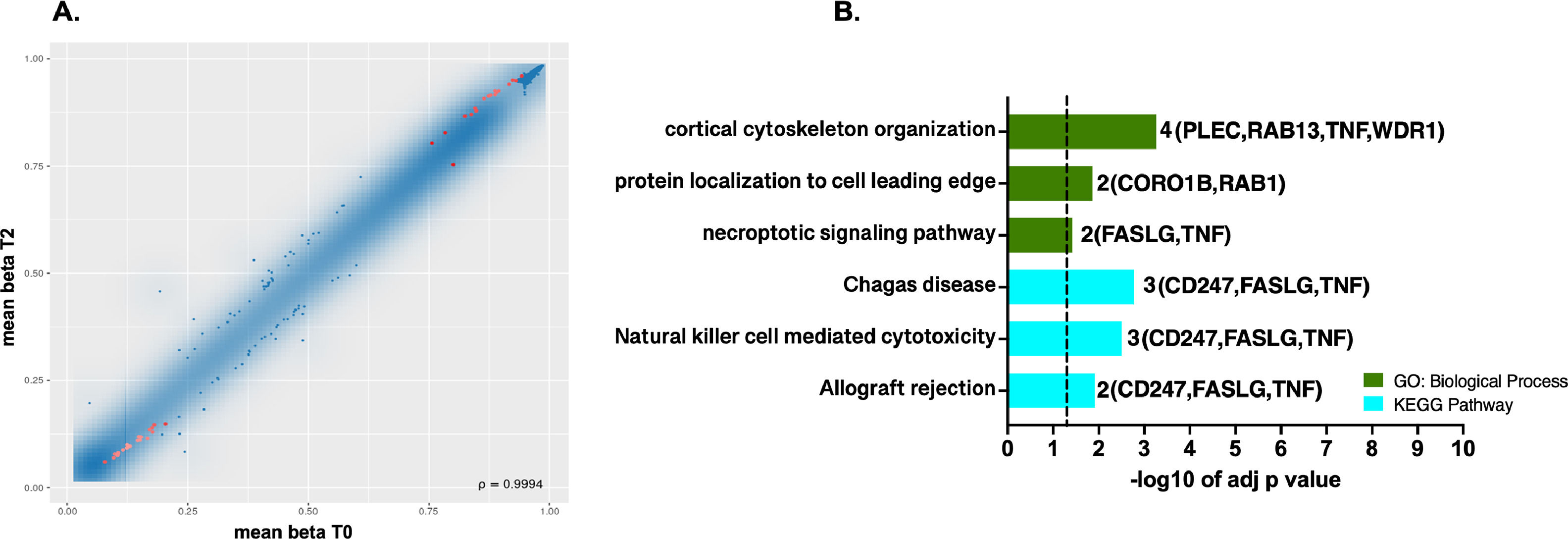
3Results3.1Differential methylation and functional enrichment analysis in the ischemic conditionsDifferences in methylation profile at site level were observed where 39 probes (17 hypermethylated / 22 hypomethylated) showed significant differential methylation between warm ischemic conditions and the baseline group (T2 vs. T0, Figs. 1A, 1B). In SM Table S2, probe's identifiers (IDs), methylation means between groups, FDR-adjusted p-values, combined rank, and methylation target sites were summarized.
 and the best-ranked sites. B. Top GO terms and pathways enriched using g: Profiler were obtained for the genes targeted by the 39 probes showing differential methylation at the CpG site level. The set of genes for GO/Pathway enriched was represented by the number at the right of each horizontal bar. The vertical dashed lines represent significant at p≤0.05.")
DNA methylation profiles at single CpG sites. A. Scatterplot of the mean beta values for the pairwise comparison T2 vs. T0 at single CpG sites. Red dots in scatterplots represent the most significantly differently methylated sites with FDR (pAdj. value <0.05) and the best-ranked sites. B. Top GO terms and pathways enriched using g: Profiler were obtained for the genes targeted by the 39 probes showing differential methylation at the CpG site level. The set of genes for GO/Pathway enriched was represented by the number at the right of each horizontal bar. The vertical dashed lines represent significant at p≤0.05.
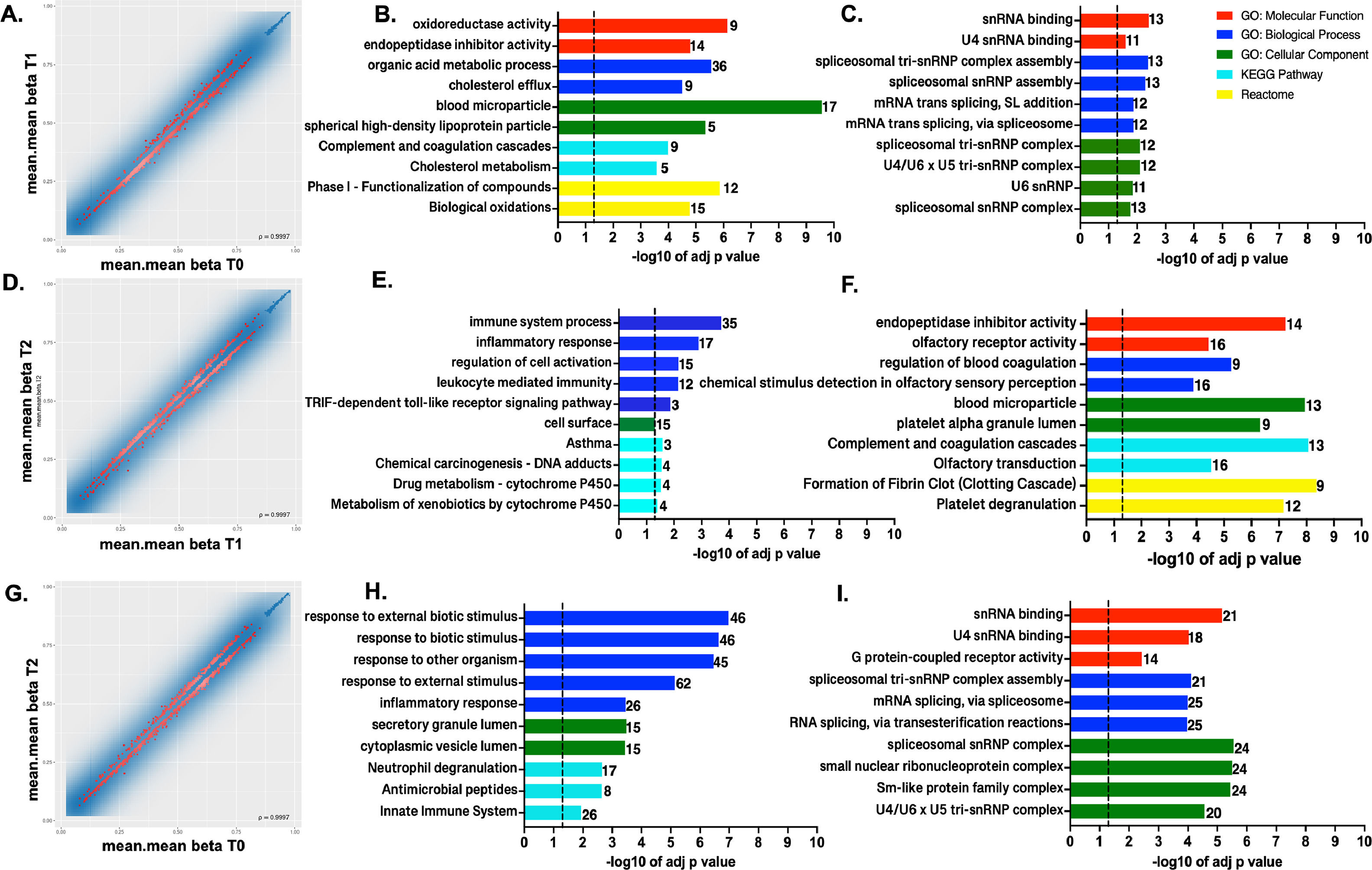
Aside from CpG sites, methylation profile for all the probes covering different genomic regions (region level: promoters, CpG islands, and tiling) was also analyzed. Based on the combined rank metrics, the top 1000 promoters exhibiting differential methylation were selected for over-representation analysis (ORA). Furthermore, these regions were grouped based on the mean difference in means (mean.mean.diff) across all sites in promoters either hypermethylated with a putative impact at the transcriptional level, repressing gene expression, or hypomethylated, up-regulated at the expression level.
In T1 vs. T0 comparison, 781 were hypomethylated and 219 promoters were hypermethylated from the 1000 top-ranked promoters (Figs. 2 A-C). In the T2 vs. T1 comparison at the promoter level, 420 were hypomethylated, while 580 were hypermethylated (Figs. 2 D-F). Lastly, there were 613 hypomethylated promoters and 387 hypermethylated when samples at T2 were compared with the T0 group (Figs. 2 G-I).
 and hypermethylation (C, F, I) at the promoter region level in the compared conditions. The set of genes for GO/Pathway enriched was represented by the number at the right of each horizontal bar. The vertical dashed lines represent significant at p≤0.05.")
DNA methylation profiles at the promoter region level. Scatterplot of the mean beta values at promoter region level for the 1000 best-ranked sites for the pairwise comparison A. T1 vs. T0, B. T2 vs. T1, and C. T2 vs. T0. Top GO terms and pathways enriched using g:Profiler for the genes showing hypomethylation (B, E, H) and hypermethylation (C, F, I) at the promoter region level in the compared conditions. The set of genes for GO/Pathway enriched was represented by the number at the right of each horizontal bar. The vertical dashed lines represent significant at p≤0.05.
First and foremost, the hypermethylated promoters were over-represented across all comparisons in GO: BP terms clustering in mRNA splicing and spliceosomal machinery assembled processes (Fig. 2 C, F and I). Furthermore, particularly in T2 vs. T1 (Fig. 2F), there were hypermethylated promoters enriched in both GO:BP and GO:CC terms involving genes with roles in blood coagulation, blood microparticle composition, related to complement and coagulation cascades, according to KEGG and REACT.
When comparing T1 vs. T0, Fig. 2C GO, REACT, and KEGG enriched terms show that the most significant hypomethylated promoters were from gene clusters involved in biological oxidations, metabolic process, cholesterol efflux, blood microparticles formation, complement and coagulation cascades, and homeostasis. Meanwhile, promoters related to innate immune response and inflammatory response were over-represented in T2 vs. T1 (Fig. 2E). Lastly, in the T2 vs. T0 comparison, hypomethylated promoters were again associated with response to external and biotic stimuli, and innate immune and inflammatory responses (Fig. 2H).
The list of the best 1000 ranked promoters with differentially methylated sites for the compared conditions is shown in SD (Tables S3, S4, and S5).
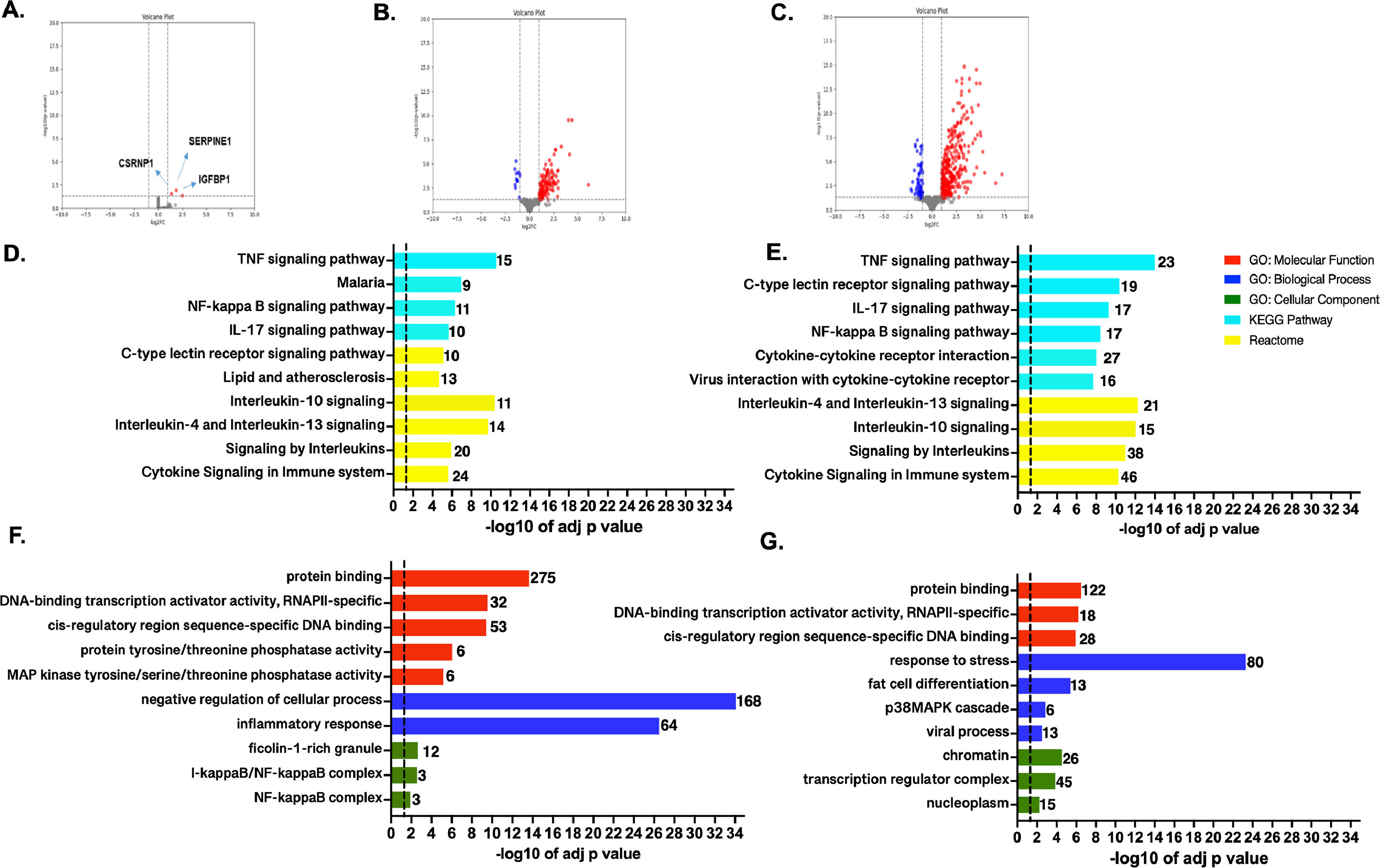
3.2Transcriptome profile and enrichment analysis in the studied ischemic conditionsTranscriptome profiles were compared among the different conditions. In T1 vs. T0 comparison, only SERPINE1, CSRNP1, and IGFBP1 showed significant upregulation (Fig. 3A). Interestingly, serine protease inhibitor, which encoded by SERPINE1, is required for fibrinolysis down-regulation and regulation of controlled degradation of blood clots [19], while CSRNP1 encodes for a protein with transcriptional activator activity [20]. Lastly, IGFBP1 is mainly expressed by the liver and circulates in plasma, prolonging the half-life of IGFs [21].
. Top GO terms and pathways enriched by GO, KEGG, and Reactome were obtained for the genes showing up-regulation on T2 vs. T1 (C and D) and T2 vs T0 (F and G). Functional enrichment was performed using g:Profiler. The set of genes for GO/Pathway enriched was represented by the number at the right of each horizontal bar. The vertical dashed lines represent significant at p≤0.05.")
Genome-wide transcriptome profile. Volcano plots for the pairwise comparisons: A. T1 vs. T0, B. T2 vs. T1, and E. T2 vs T0. Blue and red dots represent up-regulated and down-regulated genes, respectively (according to the log2FC and FDR, see Methods). Top GO terms and pathways enriched by GO, KEGG, and Reactome were obtained for the genes showing up-regulation on T2 vs. T1 (C and D) and T2 vs T0 (F and G). Functional enrichment was performed using g:Profiler. The set of genes for GO/Pathway enriched was represented by the number at the right of each horizontal bar. The vertical dashed lines represent significant at p≤0.05.
In T2 vs. T1 comparison, 139 out of the 151 DEGs were up-regulated during reperfusion with the recipient's blood (Fig. 3B and Table S6). Furthermore, based on ORA analyses the up-regulated genes were strongly associated with transcription and signaling pathways regulation, mainly through protein phosphorylation (Figs. 3 C-D). Several genes clustered in GO: BP terms were also related to stress response and activation of p38MAPK cascades. The enriched GO:CC terms belong to nuclear categories (chromatin, transcription complexes, etc.). KEGG and REACT databases, enriched terms associated with innate and adaptative immune responses, involve TNF-α, NF-KB, and several interleukins-mediated signaling pathways (IL-17, IL-4, IL-13, etc.). Otherwise, 12 down-regulated genes showed no functional enrichment. Nevertheless, top significant down-regulated genes such as BLTP2, JRK, UNC119B, DENND11, and GCN1, are involved in lipid transport, DNA-binding [22], cilium biogenesis/degradation [23], cargo adapter, guanine-nucleotide exchange, and response to amino acid starvation [24], respectively.
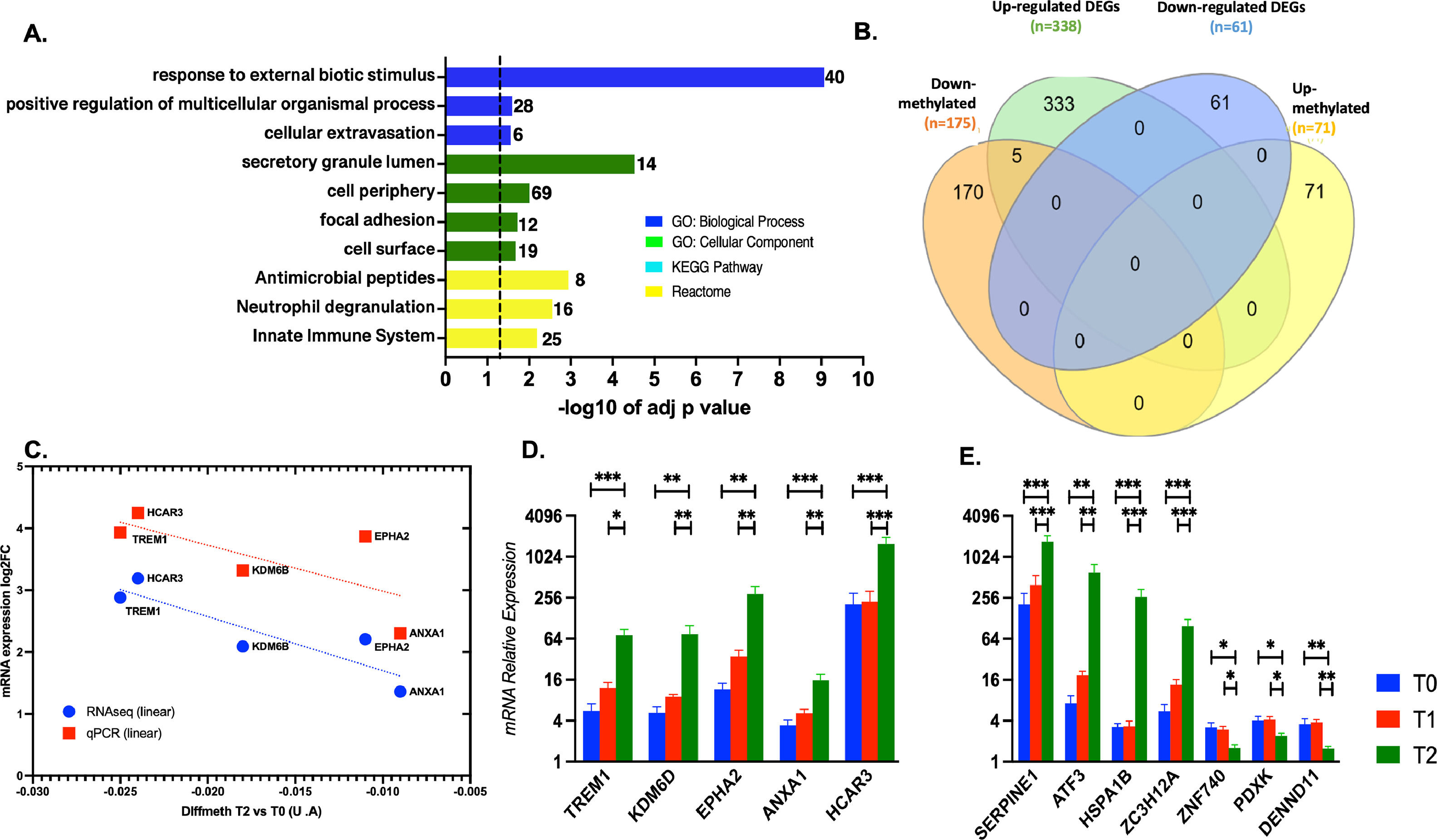
When comparing T2 vs. T0 condition, 398 DEGs were identified, most of them significantly up-regulated (n = 337), and the rest were down-regulated (n = 61) (Fig. 3E). A complete list of the DEGs, fold changes, and p-adjust values are reported (SM - table S7). Based on ORA analysis, GO:MF terms are related to negative regulation of the cellular process, inflammatory response for GO:BF terms, and transcription factors' complexes (GO:CC). Immune response was the predominant enriched term from KEGG and REACT (Figs. 3 F-G).
Enrichment of down-regulated DEGs was not obtained in any GO term by gProfiler. The trend for the 10 most significant up-and-down-regulated genes is presented in SM (Figs. S1-S2).
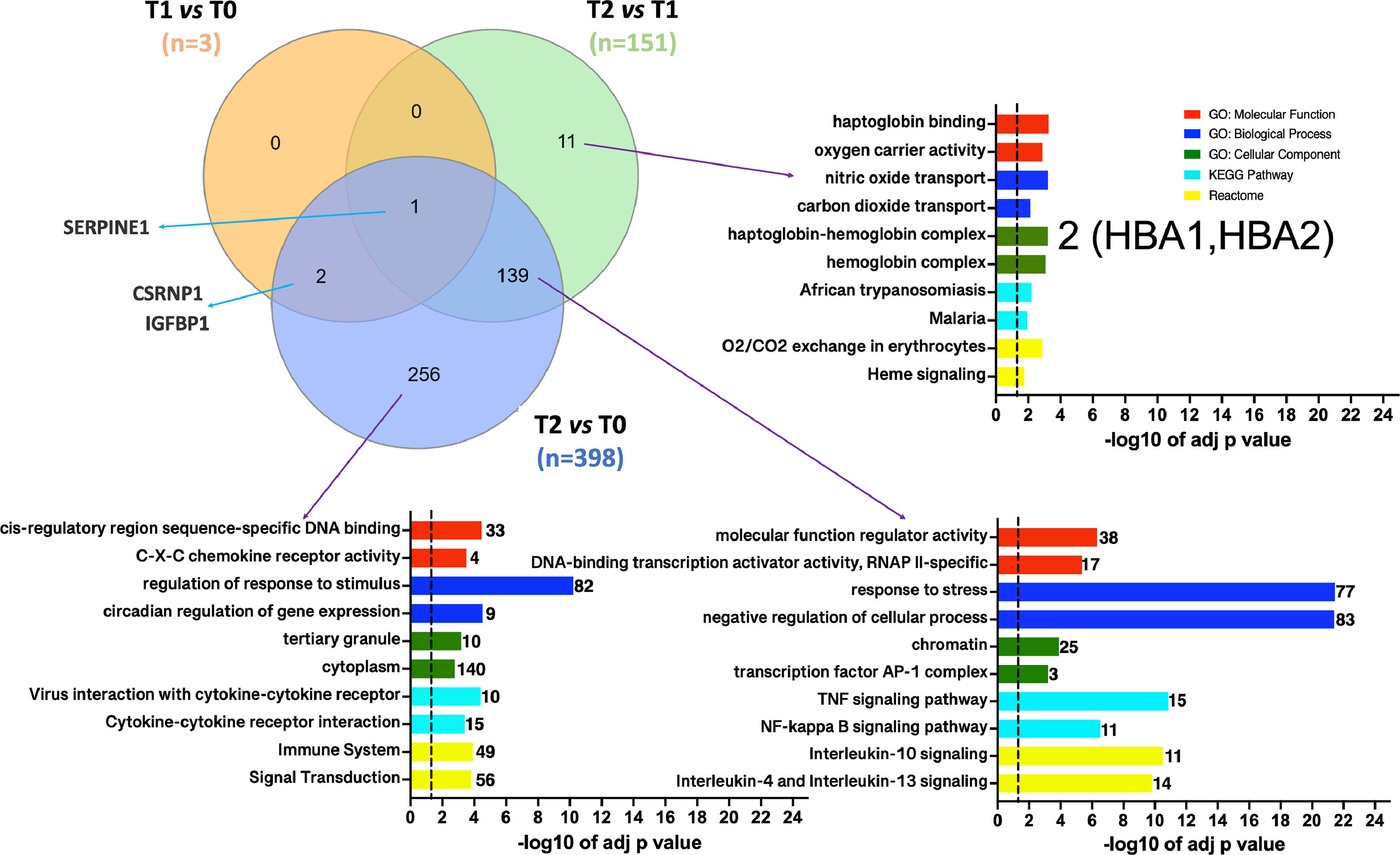
3.3Defining DEGs subsets associated with the ischemic conditionsVenn diagrams [25] were generated to investigate which DEGs were shared or unique among the different conditions. Fig. 4 illustrates that SERPINE1 is the sole DEG common across all pairwise dataset comparisons. Moreover, the diagrams identified 11 DEGs (10 up-regulated) particularly associated with the reperfusion with the recipient's blood regarding the cold preservation state. This particular set of genes should represent the response of hepatocytes to thermal or hypoxia to normoxia changes. The ten up-regulated genes were involved in oxygen/carbon dioxide transport, erythrocytes oxygen exchange, heme, and ER stress signaling pathways. Among those genes, HBA1, HBA2, and DDIT3 were enriched.

Venn diagrams depicting the overlap between DGEs in the different pairwise comparisons. T1 vs. T0 in orange, T2 vs T1 in green, and T2 vs T0 in purple. Functional enrichment was performed for indicated subsets. The set of genes for GO/Pathway enriched was represented by the number at the right of each horizontal bar. The vertical dashed lines represent significant at p≤0.05.
Moreover, the set of 139 DEGs (128 up-regulated and 11 down-regulated) common among datasets T2 vs. T1 and T2 vs. T0 were enriched. The most significant GO categories were response to stress, activation of metabolic process, and clusters participating in the inflammatory response (mediated by TNF-α, NFKB, and interleukins). Lastly, 256 DEGs (206 up-regulated and 50 downregulated) were unique to the T2 vs. T0 dataset. Ontological information was available for up-regulated genes, over-representing in GO: BP terms such as response to stimulus and circadian regulation of gene expression. Moreover, enrichment of the same signaling pathways by the shared set of 139 genes was obtained. The Venn's diagram full DEGs list is detailed in SM (tables S8-S9-S10).
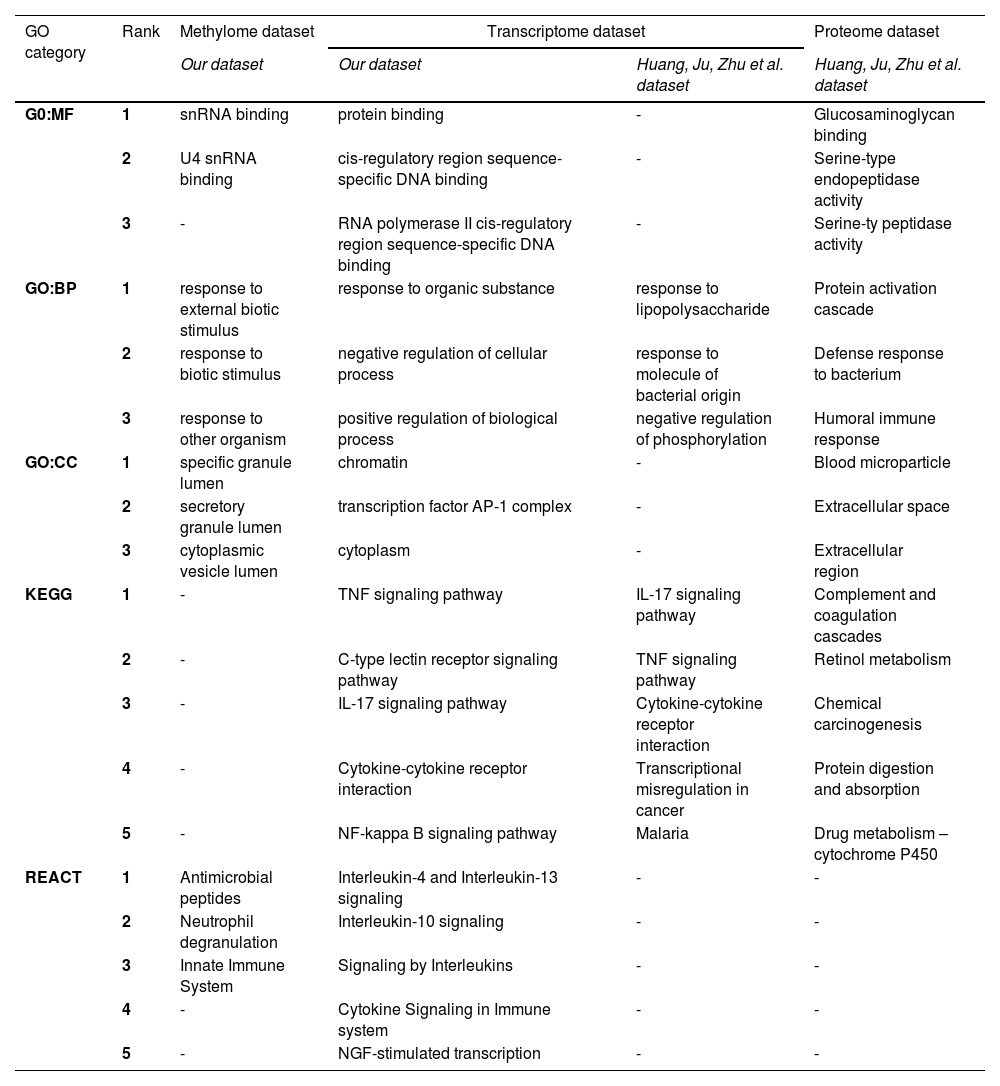
To have a complete picture of the molecular pathways playing key roles in IRI, we compared our data with the most recent information available in the literature. Table 3 summarizes the three most significant GO-enriched terms and the five most significant pathways (from KEGG and REACT) for methylome and transcriptome profiles and the information from the Huang Ju Zhu et al. study [26].
3.4mRNA expression validation by qPCR of selected most up/down regulated and hub candidates’ genesTo identify interesting genes as candidates for validation, we cross-math the dataset about differentially methylated promoters with the dataset of DEGs when T2 was compared with T0. To do this, the 1000 best-ranked promoters showing differential methylation were categorized as protein-coding or not. 175 down- and 71 up-methylated promoters were classified as protein-coding genes, and the rest were categorized as miRNAs, lnRNA, snoRNAs, etc (Table 2). As expected, the functional enrichment of the 175 down-methylated promoters was consistent with the enrichment for the total down-methylated 1000 best-ranked promoters (Fig. 5 A). Then, we performed the cross-match with the transcriptome dataset using Venn diagrams (Fig. 5 B). Our data demonstrated that five down-methylated promoters were linked to their five respective transcripts, showing expression up-regulation for the coding-protein gene. qPCR results confirmed the changes observed by transcriptomics, validating the five common genes (TREM1, KDM6D, EPHA2, ANXA1, and HCAR3). Moreover, the additional seven selected genes -SERPINE1, ATF3, HSPA1B, ZC3H12A – and -ZNF740, PDXX, and DENN1 – the 4 and 3 most up and down-regulated, respectively, were also tested by qPCR, also confirming the obtained transcriptome profile for them (Fig. 5 C-F and SM Figs. S1 and S2). In addition, it was notable that EPHA2 and ANXA1 (which were 5 and 15 folds increased during the post-reperfusion condition, respectively) were individuated as putative hub genes (with direct interaction) in a protein-protein interaction network biology analysis associated with the immune response (SM – Table S12 and Fig. S3).
Biotype categorization of the best 1000 ranked promoter when comparing T2 vs T0.
| Condition T2 vs. T0 | Methylated promoters (n = 1000) | |
|---|---|---|
| Down methylated (n = 613) | Up-methylated (n = 387) | |
| Protein coding-genes | 175 | 71 |
| miRNA, lncRNA, miscRNA, snoRNA, pseudogenes, others | 438 | 316 |
Ensembl biotype annotations were used for the classification of the differential methylated gene promoters in coding or non-coding genes at T2 vs. T0.
Comparison of enriched functions in the methylome, transcriptome, and proteome during the ischemic conditions in liver transplantation
Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome (REACT) functional enrichement analysis were performed using the g: Profiler platform. Molecular Function (GO:MF), biological process (GO:BP), and cellular component (GO:CC), as well as KEGG and REACT terms with P < 0.05. The first three and five best significant ranked enriched categories/pathways are detailed in the table for GO, REACT and KEGG respectively.
. C. Differential methylation by microarrays vs. mRNA differential expression by RNAseq and qPCR for the 5 common genes. D and E. mRNA relative expression by qPCR for the 5 common and the 7 additional selected genes at T0, T1 and T2. Data are shown as mean ± SD. Group comparison by one-way ANOVA *p < 0.05, ** p < 0.01, *** p < 0.001.")
Validation of interesting targets from methylome and transcriptome datasets. A. Functional enrichment of the 175 differential methylated promoters in T2 vs. T0. The set of genes for GO/Pathway enriched was represented by the number at the right of each horizontal bar. The vertical dashed lines represent significant at p≤0.05. B. Venn diagrams depicting the overlap between differential methylated promoters and differential expressed genes, coding for proteins (T2 vs. T0). C. Differential methylation by microarrays vs. mRNA differential expression by RNAseq and qPCR for the 5 common genes. D and E. mRNA relative expression by qPCR for the 5 common and the 7 additional selected genes at T0, T1 and T2. Data are shown as mean ± SD. Group comparison by one-way ANOVA *p < 0.05, ** p < 0.01, *** p < 0.001.
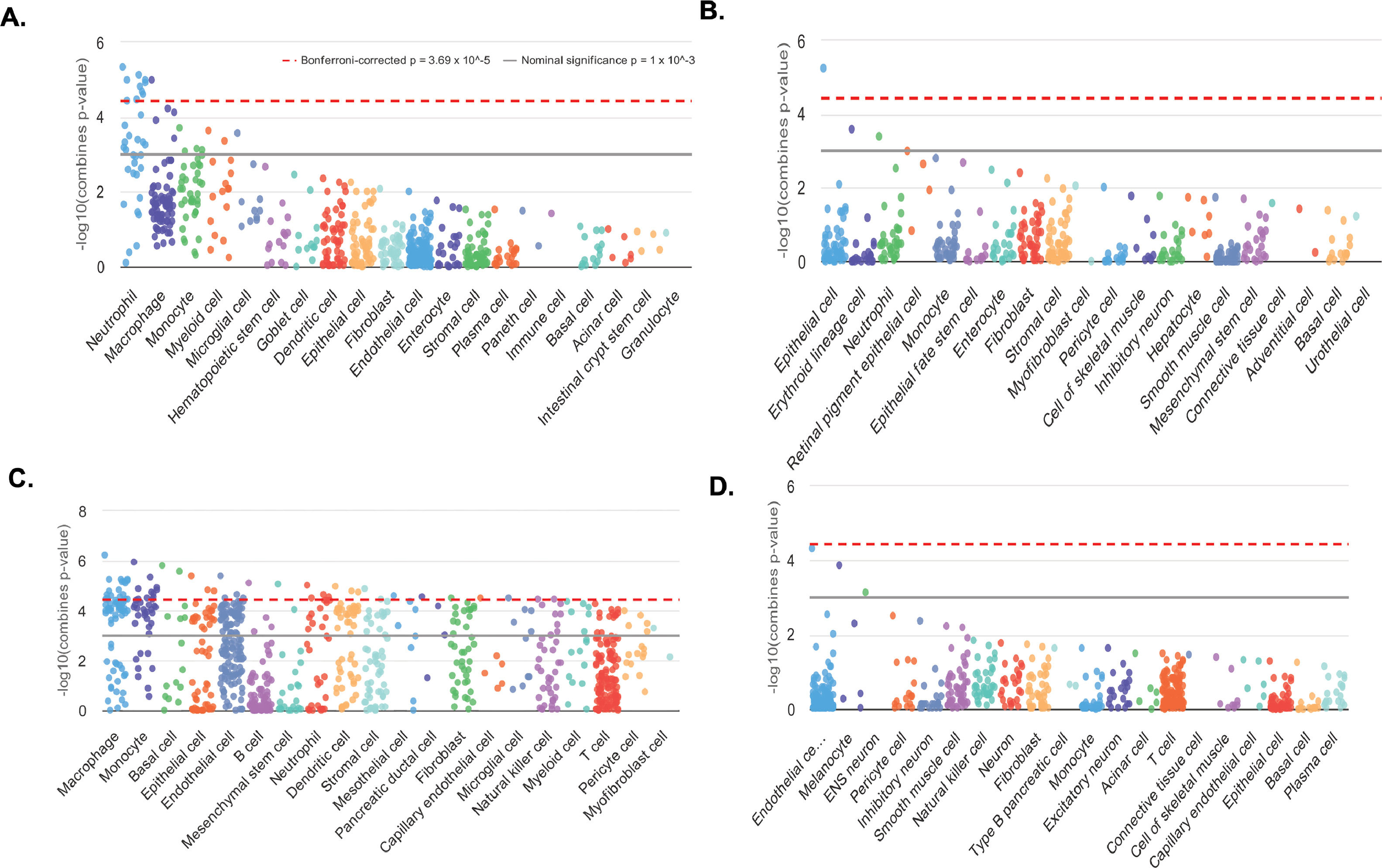
Cell type enrichments were obtained by querying WebCSEA platform for the information about up- and down-methylated promoters associated to coding-protein genes (data for 169 and 65 genes of the total 175 and 71 promoters, respectively, were available). Then, the information on 314 up- and 40 down-regulated genes (of the total 338 and 61 DEGs) enables us to obtain cell type enrichments from the transcriptome dataset. The result for the enrichment of both omics datasets were consistent demonstrating that the tissue-cell-type signature genes were indicating the presence of immune cells, being the most significant: macrophages, monocytes, neutrophils, sinusoidal endothelial and dendritic cells (Fig. 6 A-D and SM - Table S11 and Fig. S4). The most significant liver-cell-type enriched signature genes are detailed in Table S11 – SM. Interestingly, both our candidates ANXA1 and EPHA2 were part of the enriched signatures of the over-represented TC.
. T2 vs. T0 CSEA based on hypomethylated and hypermethylated promoters from coding-protein genes A. and B. respectively. T2 vs. T0 CSEA based on up- and down- differentially expressed genes C. and D. respectively. In each category of general cell types, dots represent all tissue-cell types annotated in this general cell type and descend by the order of most significant tissue-cell type in this general cell type. Y-axis is the –log10 (combined p-value) from the CSEA result. The dashed red line indicates the significant threshold (p = 3.69 × 10^-5) corrected with 1355 tissue-cell types. The solid grey line indicates the nominal significance (p = 1 × 10^-3).")
Cell-type specific enrichment analysis (CSEA). T2 vs. T0 CSEA based on hypomethylated and hypermethylated promoters from coding-protein genes A. and B. respectively. T2 vs. T0 CSEA based on up- and down- differentially expressed genes C. and D. respectively. In each category of general cell types, dots represent all tissue-cell types annotated in this general cell type and descend by the order of most significant tissue-cell type in this general cell type. Y-axis is the –log10 (combined p-value) from the CSEA result. The dashed red line indicates the significant threshold (p = 3.69 × 10^-5) corrected with 1355 tissue-cell types. The solid grey line indicates the nominal significance (p = 1 × 10^-3).
DNA methylation is a common epigenetic mechanism cells use to "switch off" genes. CpG islands (the sequences where methylation occurs) are usually near upstream transcription start sites, with increased methylation correlating with low to no transcription.
This study reveals that even relatively short periods of stress can alter the DNA methylation patterns, which subsequently was reflected in the transcriptome. The DNA methylation data at the promoter level during cold ischemia indicates shifts in biological and metabolic processes, activating genes involved in coagulation, clot formation, and energy production. Interestingly, we detected SERPINE1(PAI-1) expression upregulation, the main inhibitor of tissue-type plasminogen activator (tPA) and urokinase (uPA), reducing and preventing fibrinolysis [19,27]. SERPINE1 modulation in IRI, as we observed, has been described earlier in the literature [28–30]. LT procedures involve vascular manipulation in a complex scenario where coagulopathy may occur due to several factors (temperature changes, hemodilution, calcium and acid-base imbalance, etc.). This is particularly evident during the anhepatic phase (the time from the physical removal of the liver from the recipient to recirculation of the graft), where fibrin formation can initiate from the absence of coagulation factor synthesis and clearance of activated fibrinolytic factors. Increased levels of tPA correlate with an increase in PAI-1 has been noted; however, sometimes, the levels of PAI-1 are insufficient to avoid progression in pathological fibrinolysis [31,32].
In contrast, the DNA methylation signature obtained during reperfusion, and after cold ischemia demonstrated shifts from hypomethylation to hypermethylation at the promoter level. Interestingly, gene clusters related to coagulation cascades, blood microparticles formation, and platelet degranulation have been deactivated. This finding might help understand over-coagulation processes seen in many LT cases, which are responsible for non-technical factors related to early arterial thrombosis or changes in the intrahepatic vascular pattern observed when early CT scans are performed for other clinical reasons.
In addition, gene sets associated with the immune response were hypomethylated, indicating a presumed activation at the transcriptome level. Up-regulated genes enrichment when comparing T2 vs. T1 showed a strong stress response (80 genes) in combination with the activation of inflammatory pathways (51 genes, -TNF-α NFKB, and Interleukins-mediated signaling pathways among them). Furthermore, Venn diagrams illustrate 11 unique DEGs related to gas exchange and ER stress (including hemoglobin subunits HB1A and HB2A), which are also present in reported LT omics literature [26,33]. From this finding, we hypothesize the presence of RNA transcripts from erythrocytes and other blood cells during reperfusion since adult hepatocytes did not express hemoglobin subunits coding genes [34].
Recently studies using scRNAseq have been published; Wang L. et al. confirmed that most NK and T cell clusters were active in the TNF-α and NFKB signaling and other pathways related to inflammation and cell activation in the reperfusion stage. Moreover, B and plasma cell clusters were enriched in signaling pathways related to lymphocyte activation and adaptive immune systems [10,35]. In line with those studies, our cell-type-specific enrichment analysis individuate monocytes, macrophages, neutrophiles and LSECs as the active cells in the observed immune response. Further studies at single-cell level in human LT is imperative to understand how different cells trigger the immune and inflammatory response during IRI. It is also noteworthy that our methylome and transcriptome datasets, comparing the reperfusion condition with the baseline, reveal a configuration of hypermethylated promoters implicated in splicing events, probably to reestablish the hepatocyte's metabolic functions during normothermia. Moreover, it is evident a wider inflammatory and immune response from the methylome, where enriched hypomethylated promoter clusters categorized in terms related with response to biotic stimulus, to other organisms, neutrophil degranulation and antimicrobial peptides, and so on [36]. At the transcriptome level, the enriched GO terms and functional pathways were the same as those comparing T2 vs. T1 but with a strong extension. We confirmed expression level changes of the most up/down-regulated genes during reperfusion and also explore at mRNA level the changes of ANXA1 and EPHA2 two putative clue hub genes in the network biology of the inflammatory response. It is well known that EPHRIN receptors heterocomplex mediates bi-directional signaling in which numerous signaling pathways known to play a role in immune cell function can be activated through both ephrin “reverse” and ephrin “forward” signaling [37]. With regards to ANXA1 is released by dead neutrophils, limiting additional neutrophil migration by interacting with formyl peptide receptor 2 (FPR2) and prompting neutrophil apoptosis [38].
Our study has some limitations. Firstly, the analysis was performed in a small number of biopsies; a higher number of specimens may increase the robustness of the findings, mainly at the methylome level. Secondly, histology was performed only after cold ischemia, at the end of the engraftment but not at the end of the reperfusion phase; however, the injury by ischemia-reperfusion working under the same LT surgery protocol and performed by the same surgery team was evaluated by Suzuki and Ishak scores in the previously reported IRI proteomics analysis [39].
5ConclusionsIn conclusion, despite these limitations, our study highlights the relevance of methylation in the IRI during LT, even seeming complementary to transcriptomics analysis and providing a complete picture of the ongoing biomolecular processes. Moreover, the DNA methylation datasets generated in our study are the early literature of IRI and will be essential for other researchers' future integration of omics information. Further methylome studies in the LT outcome are expected soon to understand better how IRI or the donor-recipient environment affects the modulation of whole gene expression comparing early and late post-transplant periods. It remains to be seen if the putative epigenetic changes in the hepatocytes have a long-term effect on the phenotype of the transplanted organ. Cumulative injury dealt with by epigenetic modifications might have a lasting impact on the overall LT outcome.



