



Liver cirrhosis is a major cause of morbidity and mortality worldwide and has very limited therapeutic options. Regardless of the aetiology, hepatic fibrosis is a characteristic feature of chronic liver disease. Our knowledge regarding the pathogenesis of this scarring has grown exponentially in the past 2S years. It has now clear that this is a highly dynamic process and the long-held dogma that it is irreversible and relentlessly progressive is now being challenged. In this review, we will summarise the key pathogenic mechanisms at play and will focus on the evidence demonstrating that liver fibrosis is reversible in humans and animal models. In particular, we will examine the role of hepatic stellate cells, MMPs, TIMPs and macrophages in this process. Finally, we will discuss some of the studies aimed to therapeutically target the resolution of fibrosis and their potential for translation into a badly-needed treatment modality in the clinical setting.
Liver fibrosis is the wound healing response of the liver to a variety of insults. In clinical practice these injurious stimuli commonly take the form of chronic alcohol abuse, chronic viral infection with Hepatitis C or B, Non Alcoholic Fatty Liver Disease (NAFLD) in association with obesity and the metabolic syndrome, parasitic infection and a series of other autoimmune and metabolic conditions. If these insults remain chronically unchecked, ultimately the scarring can progress to liver cirrhosis, with the pathological hallmarks of advanced fibrosis, nodular hepatocyte regeneration, and significant architectural disruption. Such patients become susceptible to the complications of chronic liver disease, including portal hypertension, hepatocellular carcinoma and liver failure. Unfortunately current therapeutic options remain limited to management of complications and removal of the aetiological agent, barring liver transplantation for a select few. With an increasing incidence of cirrhosis and an already severe shortage of donor organs, it is clear novel therapies are vital.
The progression from liver fibrosis to cirrhosis was long thought to be inexorable. However, recent research into the mechanisms of fibrogenesis has highlighted the dynamic nature of the process and is beginning to challenge this dogma. In this concise review, we will focus on the evidence that hepatic fi-brosis is potentially reversible and highlight possible therapeutic strategies that may prove invaluable in the future.
Hepatic Myofibroblasts are the Key Scar Producing Cells in the Fibrotic LiverFollowing injury to the liver, there is cellular damage, activation of the resident inflammatory cells as well as recruitment of additional inflammatory cells. In the acute setting the extraordinary homeostatic and regenerative capacity of the liver takes over, and normal architecture is restored with only transient extracellular matrix (ECM) deposition observed.1,2
With repeated and chronic injury, inflammation is persistent or iterative and the normal wound healing response becomes aberrant with excessive deposition of ECM, primarily fibrillar collagens I and III.3,4 The principal cellular source of this ECM has been identified as the activated hepatic stellate cell (HSC). When quiescent, HSCs reside within the space of Disse and principally store retinoids. Following injury, HSCs undergo transdifferentiation into an activated myofibroblast-like phenotype, associated with loss of the retinoid droplets and expression of the filament alpha smooth muscle actin (α- SMA).5-8 They then adopt a number of pro-fibrotic features including proliferation, contractility, ECM synthesis, secretion of pro-inflammatory mediators and inhibition of matrix degradation (see below).9 Thus, they are critical in the progression of liver fibrosis.
In addition to HSCs, other sources of hepatic myofibroblasts have also been identified. Portal myofibroblasts have been defined as a distinct population, which may have enhanced significance in chronic cholestatic disease or periportal fibrosis.10-12 Bone-marrow derived myofibroblasts also contribute significantly to liver fibrosis. This interesting finding was demonstrated by studying male patients who had received a liver transplant from a female donor, with recurrent fibrosis in the graft. The authors were able to demonstrate that a significant proportion of the myofibroblasts in these livers were Y-chromosome positive, confirming their origin from circulating cells.13 This has subsequently been confirmed in murine models including tangible evidence that these cells are functional in vivo.14 Epithelial-mesenchymal transition (EMT), where hepatic myofibroblasts arise from differentiation of hepatocytes or cholangiocytes, has also been proposed in liver fibrosis,15-17 although the relative functional contribution of these cells remains to be elucidated.
Fibrosis Progression Results From a Failure in Matrix DegradationMatrix metalloproteinases (MMPs) are a group of enzymes capable of degrading different components of the ECM. Studies in whole liver of both animal models and human cirrhosis have demonstrated an increase in MMPs active against a range of ECM proteins.2,18-20 Furthermore, tissue culture studies of isolated HSCs have shown that when activated they can express a plethora of these MMPs. Early during activation, transient upregulation of MMP-13 (collagenase-3) and MMP-3 (stromelysin) is seen, followed by increases in MMP-2 (gelatinase A), MMP-9 (gelatinase B) and MMP-14 (MT-MMP).21 Whilst some of these enzymes may in fact serve to further enhance HSC activation, it is clear that there is significant matrix degrading potential already present within the fibrotic liver. So what is it that keeps these enzymes in check during progressive fibrosis?
Tissue Inhibitors of Metalloproteinases (TIMPs) are potent inhibitors of MMPs. During activation HSCs upregulate TIMP-1 and TIMP-2, even prior to expression of collagen I. This HSC derived TIMP-1, inhibits secreted MMP-2 and MMP-9 activity more than 20-fold.22,23 Subsequent studies demonstrated elevated levels of TIMP-1 and TIMP-2 in progressive fibrosis in animal models as well as explanted human liver.21,22,24 This has even lead to the proposed use of TIMP-1 as a serum marker of fibrosis.25 An elegant study Yoshiji et al confirmed a functional role for TIMP-1 in fibrosis progression. By utilising a transgenic mouse model with liver specific TIMP-1 overexpression, they demonstrated that no increase in basal fibrosis was seen but following injury the TIMP-1 overexpressing mouse had markedly increased levels of scarring.26 This combination of data has lead to the hypothesis that the overall MMP-TIMP balance is critical for fibrosis progression.
Is Human Liver Fibrosis Reversible?A careful review of the literature reveals that anecdotal evidence for the reversibility of liver fibrosis has existed for over 30 years. Subsequently, the development of effective treatments for chronic hepatitis B and hepatitis C lead to clinical trial data with liver biopsies before and after therapy. These studies convincingly demonstrated, for the first time in large cohorts of patients, that resolution of fibrosis was a real phenomenon.27-29 Similar findings have also been demonstrated in chronic liver diseases of other aetiologies including alcohol,30 autoimmune hepatitis,31,32 chronic biliary obstruction,33 hereditary haemochromatosis34 and NAFLD.35 The diversity in the range of conditions represented in this list would suggest that reversibility is potentially generic rather than disease specific and raises hope that a therapeutic intervention could be effective.
Myofibroblast Apoptosis and Matrix Degradation are Central Features of Fibrosis ResolutionRodent studies of bile duct ligation (BDL) followed by bilio-jejunal anastamosis or chronic carbon tetrachloride (CCl4) administration followed by cessation of dosing are both excellent models to study the dynamics of fibrogenesis and spontaneous resolution. In each of these situations, an established hepatic fibrosis can resolve to near normal liver architecture within 4-6 weeks.36,37 Two key histological changes are observed during this resolution phase: degradation of fibrotic matrix (see below) and a dramatic loss of the activated HSC/myofibroblast population. Closer analysis indicates that the HSCs are lost by the process of programmed cell death, or apoptosis.36,37 This finding lead to a series of in vitro studies examining the signals regulating HSC apoptosis.38 These signals include pro-apoptotic soluble mediators such as Nerve Growth Factor (NGF) which can be released from infiltrating inflammatory cells or regenerating hepatocytes.39,40 Furthermore, direct cellular contact between macrophages and activated HSCs has been demonstrated to promote HSC death.41 Intriguingly, interactions between the ECM itself and HSCs can modulate apoptosis. Binding between collagen-1 and HSCs promote activation whereas loss of integrin-media-ted contact can lead to the HSCs entering the apoptotic pathway.42 Similar findings have been demonstrated in vivo by Issa and colleagues, where a transgenic mouse expressing a non-degradable form of collagen-1 showed a failure of spontaneous fibrosis resolution and a persistence of activated HSCs in the scar following chronic CCL443 Taken together, this evidence highlights the critical importance of the myobroblast-matrix interaction in fibrosis resolution-ECM degradation will result in reduced survival/increased apoptotic signals to the HSCs, which in turn will undergo cell death leading to a reduction in ECM production.
Clearly, matrix degradation is required in addition to HSC apoptosis for adequate fibrosis resolution. Once again studies in animal models have given us critical clues into this process. Studying a series of timepoints during fibrosis resolution indicates a significant reduction in the levels of TIMP-1 and TIMP-236,44 with an associated increase in overall collagenase activity due to a switch in the MMP-TIMP balance, resulting in net ECM degradation.44 Furthermore, TIMP-1 in itself is a pro-survival signal for HSCs,45 highlighting the importance of this reduction in concentration during resolution.
Macrophages in Fibrosis ResolutionInflammatory cells, both resident and recruited, also have a key role to play in both fibrosis and resolution. Cells of the monocyte/macrophage lineage are central in this. Macrophages are capable of adopting different activation states in response to different environmental cues, including pro-and antiinflammatory and wound healing phenotypes.46 Zamara and colleagues have demonstrated that mice lacking the monocyte chemoattractant protein MCP-I, have reduced levels of hepatic inflammation following acute injury.47 Furthermore, inhibition of MCP-I with a dominant-negative mutant followed by chronic hepatic injury with dimethylnitrosamine in a rat model, clearly shows a reduction in liver macrophage infiltration and protection from fibrogenesis.48 The seminal work in this field was published by Duffield, et al.49 Using a transgenic mouse model enabling specific depletion of macrophages, they were able to mirror the above findings, showing a reduction in fibrosis and myofibroblast number by depletion during progressive injury with CCl4. Intriguingly, depletion during spontaneous resolution abrogated the improvement in fibrosis, suggesting that macrophages also have a critical involvement in matrix remodelling. This data showed, for the first time, the existence of functionally distinct subpopulations of macrophages in the liver during injury and recovery.
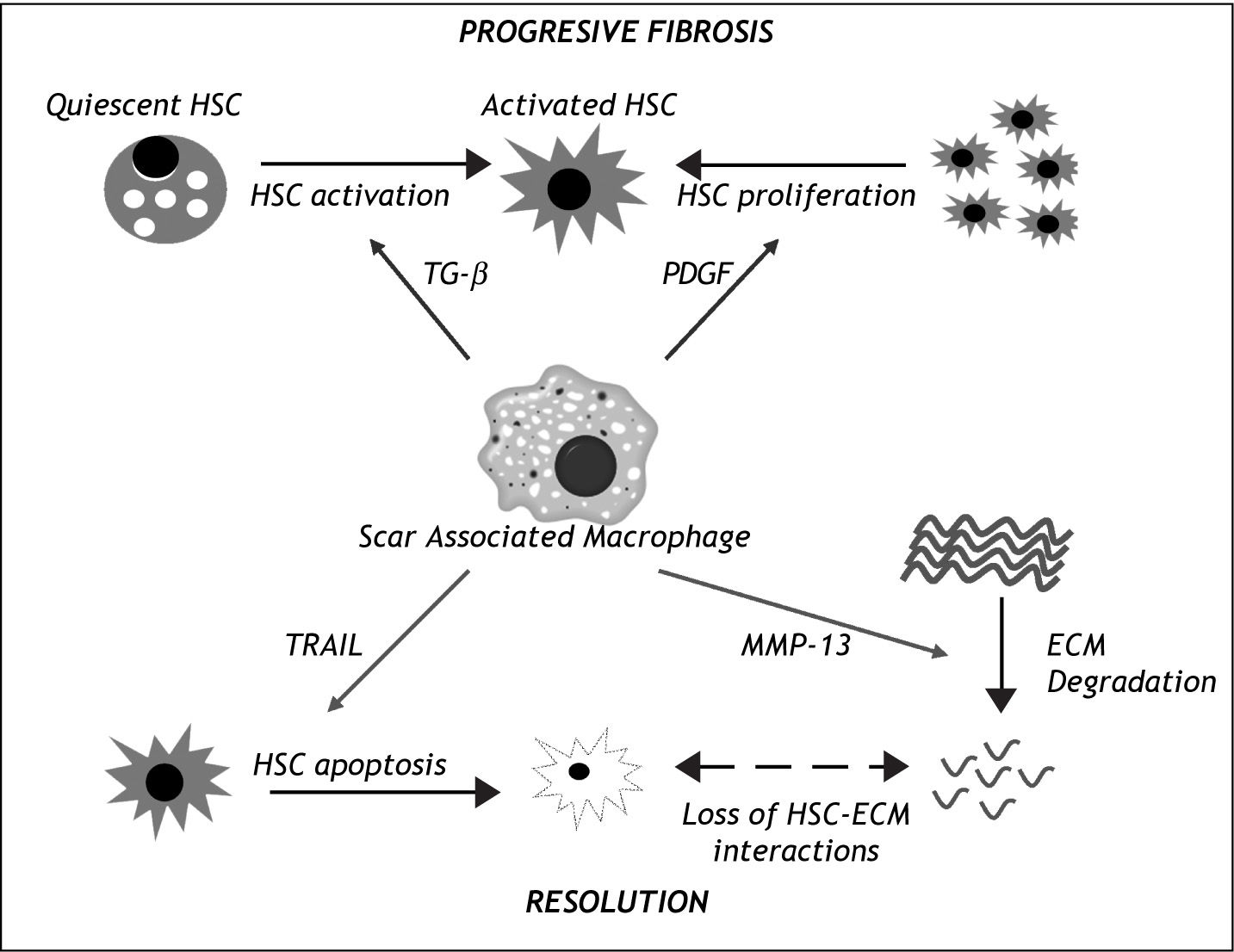
The mechanism of these macrophage driven effects has also been the subject of scrutiny (Figure I). Further analysis has revealed that following injury, there is a contribution to the hepatic macrophage pool from both liver resident Kupffer cells as well as circulating bone-marrow derived cells.49 In addition, review of the histology shows that the macrophages are located closely associated with the hepatic scar, again confirming their central role.49,50In vitro studies have demonstrated that macrophages are capable of promoting HSC activation via TGF-β production51 and increasing PDGF receptor expression,52 potentially explaining their profibrotic role. Conversely, they can also promote HSC apoptosis via expression of molecules such as TRAIL41 or MMP-9.42 Moreover, Fallowfield and colleagues showed that scar-associtated macrophages in the liver are the predominant source of MMP-13, the major rodent interstitial collagenase, and that MMP-13-deficient mice fail to resolve the scar.50 These findings raise the possibility of manipulating macrophages as a therapeutic modality.
 in liver fibrosis progression and resolution. During progressive liber fibrosis, SAMs can produce factors enhancing hepatic stellate cell (HSC) activation (e.g. TGF-β) and proliferation (e.g. Platelet Derived Growth Factor (PDGF), leading to a population of matrix-producing activated myofibroblasts. During the resolution phase, SAMs enhance loss of HSCs by apoptosis (e.g. via expression of death ligands such as TRAIL) and have a direct effect on extracellular matrix (ECM) degradation by expression of matrix metalloprotei-nases such as MMP-13. ECM degra-dtion further enhaces HSC apoptosis by loss of survival signals mediated by HSC-ESC interactions.")
The central role of Scar Associated Macrophages (SAMs) in liver fibrosis progression and resolution. During progressive liber fibrosis, SAMs can produce factors enhancing hepatic stellate cell (HSC) activation (e.g. TGF-β) and proliferation (e.g. Platelet Derived Growth Factor (PDGF), leading to a population of matrix-producing activated myofibroblasts. During the resolution phase, SAMs enhance loss of HSCs by apoptosis (e.g. via expression of death ligands such as TRAIL) and have a direct effect on extracellular matrix (ECM) degradation by expression of matrix metalloprotei-nases such as MMP-13. ECM degra-dtion further enhaces HSC apoptosis by loss of survival signals mediated by HSC-ESC interactions.
The relative contribution of other cells of the innate and adaptive immune system in liver fibrosis is less well established, although a complex interplay clearly exists.1 Inhibition of neutrophil recruitment, using either anti-neutrophil serum53 or CXCR2 knockout mice54 seems to have minimal effect on fibrosis. In a B Cell deficient mouse, however, there is reduced liver fibrosis in response to chronic CCL4 administration, while no difference was seen in T cell deficient mice by this group.55 A proposed mechanism for this finding is that there may be enhanced clearance of apoptotic hepatocytes by macrophages.56 On the other hand, B cell deficient mice show increased hepatic fibrosis in response to chronic schistosoma infection57 whilst adoptive transfer of CD8+ve T Cells from CCl4 treated mice into immunodeficient SCID mice causes activation of HSCs.58 Such findings demonstrate the difficulty in unpicking the key mechanisms involved and it is certain that disease-or model-specific factors will be at play. There also remains a lack of mechanistic data on the relative contribution of these cell types during the resolution from fibrosis.
What Renders Fibrosis Irreversible?Having described some of the evidence confirming the reversibility of liver fibrosis in human and animal models and the key mechanistic events in this process, what remains uncertain is whether advanced cirrhosis can return to normal liver architecture or whether there is a “point of no return”. It is important to note that cirrhosis is not simply advanced fibrosis, but also encompasses abnormal hepatocyte regeneration, architectural disturbance and vascular changes. Once again experimental models have given us insight into this key question. Issa et al induced cirrhosis in rats by 12 weeks of CCL4 administration, followed by a protracted recovery period of up to 1 year.44 After 12 weeks of injury, micronodules were clearly visible surrounded by dense mature fibrotic septa. Following recovery, significant remodelling was observed, primarily in the freshly formed fibrosis immediately adjacent to the nodules. The more mature scars, however, failed to resolve completely even after a prolonged period. By comparing the reversible with the potentially irreversible scars, some of the features leading to “irreversibility” could be defined. Principally, the irreversible septae contained greater quantities of the ECM component elastin. Furthermore, tissue transglutaminase mediated matrix cross-linking could be demonstrated within the irreversible scars.44 This would suggest the possibility that the more mature scar contains cross-linked matrix rendering it resistant to proteolytic degradation, similar to fibrosis in other organ systems.59 The other distinguishing feature of the irreversible scars was that they were relatively paucicellular, containing fewer active myofibroblasts and macrophages than their reversible counterparts.44 This again could be critical, the implication being that the absence of celis within the scar leads to reduced delivery of matrix degrading enzymes to the area.
Similar evidence in human liver disease is still lacking. However, a study by Wanless et al looking at explanted cirrhotic human livers, did indicate partial regression even from advanced micronodular cirrhosis.60 Strikingly, the histological patterns were remarkably similar to that described in the rat model,44 suggesting common mechanisms might be prevalent.
Targeted Therapies in Fibrogenesis and ResolutionAs described there is extensive data on the reversibility of fibrosis and at least in part of cirrhosis. This raises hopes that an effective anti-fibrotic therapy can soon be translated into the clinic. As our understanding of the pathogenic mechanisms has evolved, numerous researchers have attempted to target different fibrogenic pathways. A review of the literature would reveal hundreds of such anti-fibrotic interventions in animal models. These can broadly be divided into 3 main categories:
- 1.
Targeting the injurious stimulus or the inflammatory process ultimately leading to myofibroblast activation and ECM deposition
- 2.
Reducing myofibroblast activation and promoting myofibroblast loss by apoptosis
- 3.
Modulating the MMP-TIMP balance to favour ECM degradation
Given the importance of the resolution phase because patients invariably present to medical attention with established disease, this review will now focus on the latter 2 aspects.
Various molecules have been shown to reduce the pro-fibrotic nature of HSCs. One particular example is relaxin. This hormone is normally produced by the corpus luteum during the 3rd trimester of pregnancy and acts to soften the symphysis pubis. Studies using relaxin on activated HSCs have shown that it modulates TIMP and collagen expression in vitro and that it is anti-fibrotic in vivo,61 suggesting that recombinant relaxin or synthetic analogues may have a therapeutic role. The peroxisome prolife-rator-activated receptors (PPAR) also have an important role in regulating HSC behaviour. During HSC activation a marked reduction of PPARy expression is seen and synthetic PPARy agonists reduce HSC proliferation, chemotaxis and collagen-1 expression.62-64 These drugs are already widely used in the management of type 2 diabetes and the in vitro findings tally nicely with the recently published clinical trials in NAFLD, demonstrating histological improvement in the liver following treatment.65-67 Angiotensin II also has an activating effect on HSCs, probably mediated via TGF-0 expression, and blockade of the renin-angiotensin system seems to ameliorate experimental liver fibrosis68-70 as well as potentially having benefits in human disease.71 This correlates well with the reported effects on cardiac and renal fibrosis.72,73
Given critical nature of HSC apoptosis in fibrosis resolution, several authors have attempted to stimulate this as an anti-fibrotic therapy. As discussed above activated HSCs respond to a variety of apoptotic signals and express several death receptors including p75, FAS and TNF-α, in addition to the critical interaction with ECM.2 Attempts to specifically target this have been made, but are limited by potential off target effects due to the pleotropic nature of these molecules. An observation that activated HSCs persistently express high levels of NF-κB, a pro-inflammatory transcription factor, and that this confers protection from HSC apoptosis has lead to additional strategies to target this process.74 Sulphasalazine, a widely used anti-inflammatory drug, has been shown to induce HSC apoptosis in vitro and in vivo via an NF-κB dependent mechanism and accelerates fibrosis resolution in a rat model.75 Gliotoxin, a fungal metabolite, also induces HSC apoptosis, at least in part due to NF-κB inhibition, and thus has anti-fibrotic effects.76 This also had off target effects causing depletion of hepatic macrophages. To circumvent this problem authors have used an antibody raised against myofibroblasts to target the gliotoxin to HSCs and gain a much more specific effect.77 This certainly represents a significant advance, and similar methods may be applied more widely in the future.
Having described the importance of the MMP-TIMP balance in the progression and resolution of fibrosis, not surprisingly various studies have attempted to target this. Initially attempts were made to increase hepatic MMP levels. Siller-Lopez and colleagues administered adenovirus causing hepatic overexpression of MMP-8 (neutrophil collagenase) and showed that this significantly reduced fibrosis following BDL in rats.78 TIMP inhibition as a therapeutic avenue has also received some attention. A TIMP-I neutralising antibody resulted in reduced progression in a rat CCL4 model.79 Roeb and colleagues used a particularly novel method of TIMP inhibition. They developed a series of MMP-9 mutants, which lacked MMP gelatinolytic activity, but retained their TIMP binding properties. These mutants were able to inhibit TIMP-I effects in vitro and in vivo and reduced fibrosis in a murine model.80,81 This strategy would once again have the advantage of minimising off-target effects, by specifically having action in areas where TIMP-I would be upre-gulated during fibrogenesis.
Clearly a full discussion of the myriad of potential anti-fibrotic therapies that have been tried in experimental models is beyond the scope of this article. However, what is clear from the selected examples, is that our greater understanding of the pathogenesis has enabled specific and targetted therapies to be developed. The challenge remains in the translation of these to the clinical setting, in particular the avoidance of off-target effects which could potentially cause adverse events in humans.
The Potential of Cell TherapyAs discussed above, a cardinal feature of the irreversible scar is its hypocellularity. Many groups are now exploring the use of cellular therapy for liver fibrosis. This could potentially have the benefit of seeding the irreversible scar, enabling modification of the local microenvironment and possible degradation. A variety of cell types have been used for this purpose, with a focus on a stem cell fractions. Sakaida and colleagues, for example, used adoptive transfer of GFP-expressing bone marrow cell fraction into mice during progressive CCL4 induced injury. They were able to demonstrate a reduction in fibrosis, and also that the injected cells localised around the scars and co-expressed MMP-9.82 The implication of this is that they may become pro-resolution scar-associated macrophages. It has also been proposed that injected stem cell fractions could differentiate and contribute to the regenerating hepato-cyte pool, although this remains controversial.83 Clinical trials of cell therapy have been conducted-whilst these are generally small, non-randomised and do not show unequivocal benefit, they do offer some hope for the future.84
A criticism of the cell therapy strategy would be that following adoptive transfer, the cells will encounter the existing microenvironment in the liver, meaning their effect could be abrogated by phenotypic reprogramming according to the local conditions. A way to circumvent this problem would be to modify the cells ex vivo into an anti-inflammatory or proresolution phenotype, prior to delivery. Similar experiments have been effective in animal models of renal injury, using macrophages either stimulated with cytokines85 or virally transduced to express anti-inflammatory cytokines86,87 prior to adoptive transfer. Studies such as these are still lacking in liver fibrosis models.
ConclusionOur understanding of the pathogenesis of hepatic fibrosis has progressed exponentially in the past 30 years. In particular, we now have cogent and consistent evidence that fibrosis (and to a lesser degree advanced cirrhosis) is potentially reversible in humans and animal models, and have an insight into the critical molecular and cellular pathways involved. We have also gained knowledge of some of the features preventing resolution of mature scars. Utilising these findings, several novel therapies have been trialled in reproducible animal models of liver disease. What remains elusive is effective translation into the human setting. In the future, cellular therapies or treatments targeting multiple points in the complex fibrogenic pathway may help to bridge this gap.
Abbreviations- •
ECM: Extracellular matrix
- •
HSC: Hepatic stellate cell
- •
MMP: Matrix metalloproteinase
- •
TIMP: Tissue Inhibitor of metalloproteinases
- •
TRAIL: TNF-related apoptosis inducing ligand
Prakash Ramachandran is funded by The Wellcome Trust.
John P Iredale is consultan for GE Healhcare and is funded by The Wellcome Trust, Medical Research Council United Kingdom and the Childrens Liver Disease Foundation.



