




Edited by: Sonia Roman
More infoObesity is a risk factor for developing nonalcoholic fatty liver disease (NAFLD), and the associated molecular mechanisms could be targeted with nutrient-based strategies. Therefore, it is necessary to review the current mechanisms to propose further treatments. Obesity facilitates the onset of insulin resistance, lipidic abnormalities, hepatic fat accumulation, lipid peroxidation, mitochondrial dysfunction, excessive reactive oxygen species (ROS) production, and inflammation, all related to further steatosis progression and fibrosis. Microbiota alterations can also influence liver disease by the translocation of pathogenic bacteria, energy extraction from short chain fatty acids (SCFAs), intestinal suppression of the expression of fasting-induced adipose factor (FIAF), reduction of bile acids, and altered choline metabolism. There are also genetic polymorphisms in metabolic proteins that predispose to a higher risk of liver diseases, such as those found in the patatin-like phospholipase domain-containing 3 (PNPLA3), transmembrane 6 superfamily member 2 (TM6SF2), membrane-bound O-acyltransferase domain-containing 7 (MBOAT7) or also known as lysophosphatidylinositol acyltransferase 1 (LPIAT1), transmembrane channel-like 4 genes (TMC4), fat mass and obesity-associated protein (FTO), the b Klotho (KLB) and carboxylesterase (CES1). No clear dietary guidelines target all mechanisms related to NAFLD development and progression. However, energy and carbohydrate intake restriction, regular physical exercise, supplementation of antioxidants, and restoration of gut microbiota seem to have beneficial effects on the new proposed features of NAFLD.
Obesity is highly associated with the development of nonalcoholic fatty liver disease (NAFLD) [1]. Globally, the prevalence of NAFLD is about 24% [2] and about 10% in developing countries [3]. It was recently estimated that the prevalence of NAFLD in Latin America could be around 24% [4]. From an epidemiological viewpoint, this high prevalence reflects the nutrition transition occurring in many nations worldwide, including Latin America, where many young individuals are obese and at risk for NAFLD at the early stages [5]. Thus, prevention strategies based on the genetic and environmental factors prevailing in the community are warranted.
NAFLD includes liver abnormalities such as simple steatosis, nonalcoholic steatohepatitis (NASH), cirrhosis, and hepatic carcinoma [6,7]. Between 10% and 29% of NASH cases may evolve into cirrhosis, and 4%-27% may develop hepatocellular carcinoma. However, obesity features could be influencing a nonreported higher prevalence. In the present review, we intend to summarize the updated molecular mechanisms of liver damage associated with obesity and review which foods and bioactive compounds can influence these mechanisms. This review could help to design nutrient-based strategies to prevent and treat NAFLD.
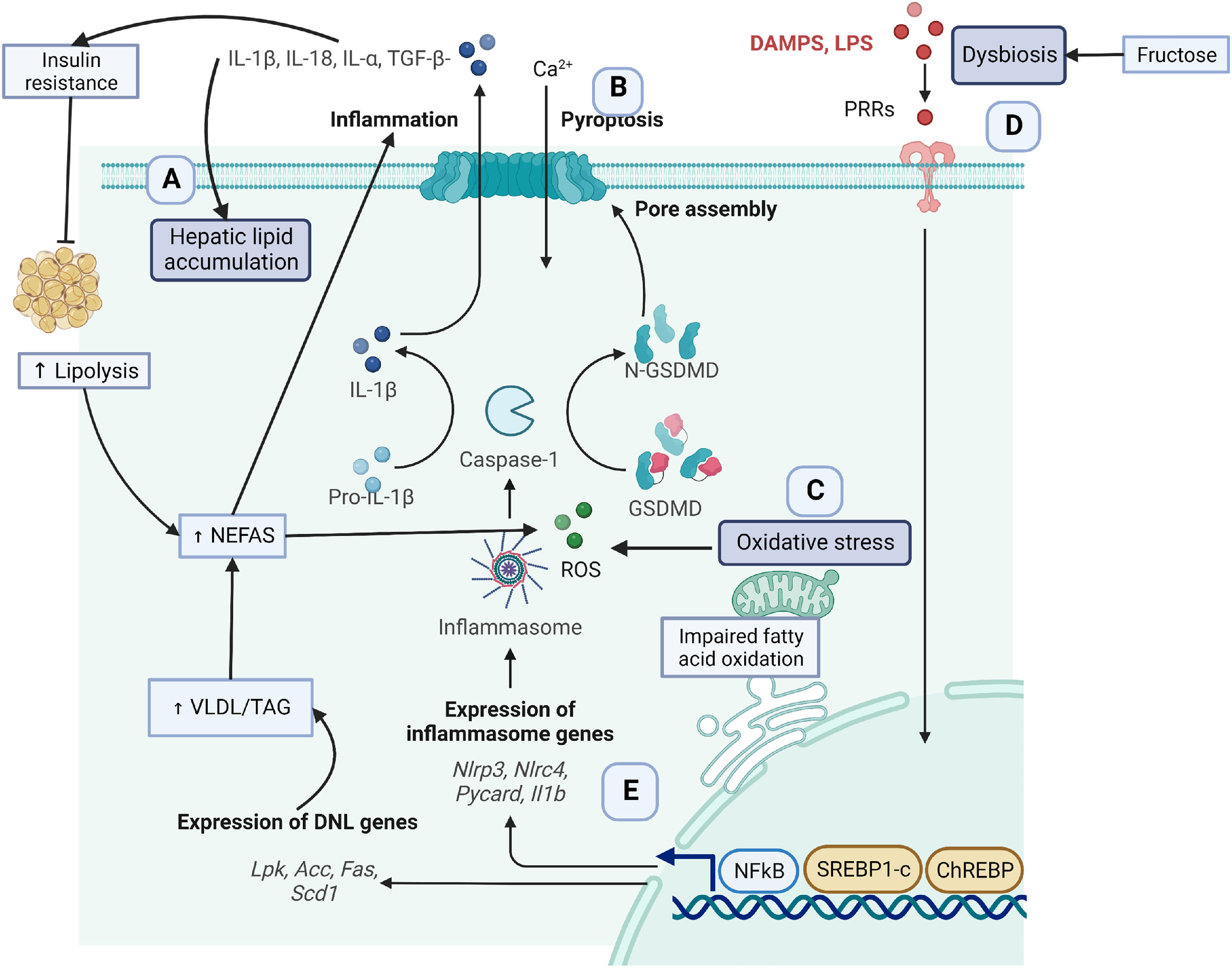
2Key obesity-associated fatty liver disease molecular mechanismsGenetic, metabolic, and lifestyle factors seem to be related to the spectrum of obesity-related liver disease [1]. In the context of obesity, excessive fat deposition seems to drive the progression of NAFLD features [8,9]. Some mechanisms contributing to excessive adipose tissue are insulin resistance, lipidic abnormalities, hepatic fat accumulation, lipid peroxidation, mitochondrial dysfunction, excessive production of reactive oxygen species (ROS), and inflammation [10,11] (Fig. 1). The following sections will detail the molecular mechanisms involved in these processes as understood to date.
 hepatic fat accumulation, (B) Inflammation, (C) Oxidative stress and mitochondrial dysfunction, (D) Dysbiosis and (E) Genetics.")
Obesity, defined as excess body fat, enables hepatic uptake of fatty acids from adipose tissue and diet. In this sense, it has been recognized that the chronic consumption of the westernized diet is hepatopathogenic not only because of its high energy content [5] but also because of the high content of sugars (fructose), saturated fatty acids, trans fatty acids, and cholesterol that induces liver fat accumulation [12,13]. However, diet only contributes 15% of hepatic lipid accumulation [14]. As illustrated in Fig. 1, (section A), about 60% of lipid accumulation is related to excessive uptake of non-esterified fatty acids (NEFAs) from adipose tissue lipolysis in insulin resistance models [15]. In turn, NEFAS induces the TLR4/PI3K/AKT inflammatory pathway contributing to more insulin resistance [16]. Insulin resistance promotes de novo lipogenesis (DNL), contributing to another 26% of lipids [14]. Insulin resistance induces the overexpression of the cellular differentiation 36 (CD36) scavenger receptor and the translocation of the sterol regulatory element-binding protein 1 (SREBP1) transcription factor to the nucleus to activate hepatic DNL [17].
In addition, fructose intake contributes to DNL in an insulin resistance environment [18]. Once the liver uptakes fatty acids, these particles can be oxidized by β-oxidation in the mitochondria and peroxisomes and by the endoplasmic reticulum via α-oxidation and ω-oxidation [19,20]. Impairments in fatty acid oxidation capacity (FAO) also contribute to lipid accumulation. In NASH human samples, the FAO capacity and β-oxidation are reduced by ∼40%-50% compared with control samples, and hepatic mitochondrial ROS is increased [21].
Alterations in VLDL transport are another way to increase lipid accumulation in the liver. Excessive energy intake and fructose induce a higher hepatic triglyceride transport via VLDL secretion [18]. Fructose also contributes to other alterations such as glucotoxicity and lipotoxicity [22]. However, in severe steatosis, the export and secretion of VLDL particles seem to be affected
2.2The inflammasomeLipidic precursors and other metabolites activate an inflammatory process related to the progression of steatosis and fibrosis [23]. One of the main inflammatory events is the release of danger-associated molecular patterns (DAMPs) from damaged hepatocytes [24]. Adenosine triphosphate (ATP) is one of the most studied DAMPs released to the extracellular space, achieving very high concentrations facilitating the activation of purinergic receptors P2X and P2Y. Specifically, the inflammasome facilitates a type of cell death called pyroptosis [25], Fig. 1 (section B). In the cytoplasm, the liver expresses different types of inflammasomes that are released to the extracellular space contributing to inflammation.
Inflammasomes are a multi-protein complex , including a sensory molecule such as nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), an adaptor protein ASC (apoptosis-associated speck-like protein containing CARD), and an effector molecule pro-caspase 1. DAMPs and pathogen-associated molecular patterns (PAMPs) are recognized by the pattern recognition receptors (PRRs), activating the NK-κB transcription factor that induces overexpression of NLRP3 [26,27]. ROS binds to NLRP3, which recruits ASC, and in turn, caspase 1 is activated. This process induces gasdermin D (GSDMD) to form cell pores to facilitate the proteolytic cleavage and release of proinflammatory cytokines, such as IL-1β, IL-18, IL-1alpha, and transforming-growth factor (TGF)-β [24]. These cytokines facilitate lipid accumulation, activation of fibrogenesis in hepatic stellate cells, and collagen production leading to tissue fibrosis and hepatic insulin resistance [28].
2.3Oxidative stress and mitochondrial dysfunctionIn the early stages of NASH, the liver has a higher mitochondrial fatty acid oxidation until it reaches its maximal limit for lipid accumulation. Next, free fatty acids induce mitochondria dysfunction. As a consequence, there is a decrease in mitochondrial respiration, and an increase in reactive oxygen species (ROS) [29], Fig. 1, Section C. Important structural and metabolic alterations in the mitochondria membrane facilitate liver disease progression [30,31]. A recent study in NAFLD-induced mice found important alterations in hepatic mitochondrial lipid profile where increases in lysophosphatidylcholines, fatty acids, diglycerides, and triglycerides and increases in ROS were experienced [32].
3Dysbiosis and liver damageIntestinal human microbiota confers multiple beneficial properties to the host [33,34]. However, whenever there is an increase in the abundance of pathogenic species in contrast to beneficial species, there is a high risk of developing chronic diseases, Fig. 1 (section D). For example, in obese patients with simple steatosis, there is a greater abundance of pathogenic bacteria such as Lachnospiraceae, Dorea, Robinsoniella, and Roseburia, which produce volatile organic compounds related to liver damage [35]. Moreover, patients with NASH present a reduction in beneficial bacteria, including Faecalibacterium bacteria, and an increment of pathogenic bacteria such as Parabacteroides, Allisonella, and Escherichia, which affect the integrity of the intestinal barrier [36]. Escherichia coli is known to produce ethanol and disrupt the integrity of the intestinal barrier resulting in upregulation of alcohol metabolism enzymes triggering liver inflammation. Furthermore, patients with liver cirrhosis significantly reduce the proportion of Bacteroidetes, while Proteobacteria and Fusobacteria are abundant. In addition, the Streptococcaceae family correlates with the severity of the disease. In addition, patients with decompensated cirrhosis due to hepatitis B infection present a higher reduction in Bifidobacteria and lactic acid-producing bacteria and a higher increment of Enterococcus faecalis compared to asymptomatic carriers and healthy controls [37].
Unhealthy food patterns rich in saturated fats, sugars, and lack of fiber, fruits, and vegetables alter the microbiota profile, contributing to the development of multiple diseases [38]. For instance, excessive consumption of fructose available in breakfast cereals, sweet beverages, and fast food is directly associated with dysbiosis by compromising the integrity of the intestinal mucosa, allowing the passage of toxins, proinflammatory cytokines, and pathogenic bacteria into the circulation. Fructose affects bacteria diversity promoting an increment in the Firmicutes/Bacteroidetes ratio [22]. Fructose-derived metabolites such as glucose, glycerol, uric acid, SCFAs, glutamic acid, glutamine, and alanine serve as lipogenic substrates; therefore, fructose stimulates DNL as previously discussed [22].
Furthermore, intestinal epithelium integrity loss allows lipopolysaccharides (LPS) and toxins to be translocated through the portal vein. LPS and toxins are recognized by Toll-like receptors (TLR4-TLR9), which regulate the innate immune response in liver cells (Kupffer, stellar). LPS then activates the molecule signaling of MyD88 in B cells which in turn favors the translocation of NF-κB, activating the expression of proinflammatory genes, leading to a higher secretion of cytokines (TNFα, IL-1β), recruitment of neutrophils, chemokines (IFN-β) by macrophages [39–41]. These aberrant results lead to liver damage by promoting DNL, hepatic accumulation of triglycerides, insulin resistance, apoptosis, and fibrogenesis. Furthermore, it has been identified that liver-specific toll-like receptors (TLRs) can recognize bacterial compounds such as TLR4, TLR5, and TLR9 [42,43].
Bacterial fermentation of dietary polysaccharides produces short chain fatty acids (SCFAs) such as acetate, butyrate, and propionate. These SCFAs contribute 70% of the intestinal bacterial energy. SCFAs can bind to G protein-coupled receptors such as GPR43/GPR41 in the colon and adipocytes. GPR41 activates peptide YY (PYY) and glucagon-like peptide (GLP-I), reducing appetite. However, in response to a signal of excess energy, there is an increase in lipogenic enzyme activity, and a reduction of fatty acid oxidation, because insulin sensitivity in adipose tissue is blocked, and it favors lipid accumulation in the liver [44]. Specifically, acetate and propionate have been implicated in hepatic lipogenesis (cholesterol and triacylglycerides) and gluconeogenesis [45,46].
In contrast, butyrate might have protective properties against NASH and cirrhosis progression by reducing the expression of fibrogenic genes [47]. For instance, it recognized that butyrate is necessary for activating the fasting-induced adipose factor (FIAF), an inhibitor of lipoprotein lipase (LPL). When FIAF is suppressed, LPL is activated, which leads to greater incorporation of fatty acids into the cell and the accumulation of triglycerides in adipose tissue [48,49]. It also facilitates the transcription of carbohydrate-responsive element-binding protein (ChREBP) and SREBP1, facilitating triglyceride accumulation in the liver [42]. This effect seems to be reduced with the administration of butyrate [50].
Dysbiosis also seems to interfere with bile acid homeostasis, which helps emulsify dietary lipids [51]. Intestinal microbiota transforms primary bile acids into secondary bile acids (deoxycholic acid (DCA), ursodeoxycholic (UDCA), and lithocholic acid (LCA)) that have antimicrobial and cytotoxicity properties. Bile acids activate the farnesoid X receptor (FXR) and G protein-coupled receptor (TGR5), which regulate bile acid production. However, dysbiosis favors the reduction of FXR and fails to inhibit NF-kB and prevent inflammation [42]. As per alterations of the choline metabolism, it has been found that intestinal microbiota converts dietary phosphatidylcholine into choline. At the same time, the abundance of pathogens (Firmicutes and Proteobacteria) favors the conversion to trimethylamine-N-oxide (TMAO), which is a toxic product associated with reduced clearance of VLDL from the liver, thus causing steatosis [52–54].
4Genetic factors related to liver disease during obesityGenetic variants in lipid metabolism genes are currently being studied to decipher their role in the predisposition toward liver disease, Fig. 1, (section E). One of the most studied genes is patatin-like phospholipase domain-containing protein 3 (PNPLA3), which encodes a lipase involved in hepatocellular lipid remodeling and retinol metabolism at the cytoplasmic level in hepatic stellate cells. The SREBP1 upregulates its expression under higher glucose conditions. In the case of the PNPLA3 rs738409, C>G polymorphism, the 148M variant in the form of GG genotype correlates with the highest protein expression, inhibition of lipases, and in turn, favoring larger hepatic lipid droplets and fibrosis [55,56]. This 148M variant regulates the hepatic triglyceride content, and this effect seems to be induced by dietary intake. For instance, studies have shown that carriers of the risk G allele had a higher intake of omega-6/omega-3 ratio which contributes to increased liver steatosis versus the CC genotypes [57]. Similarly, consuming arachidonic acid from meat origins, carbohydrates, and sugar is also related to steatosis and fibrosis. In contrast, a higher intake of n-3 polyunsaturated fatty acids (PUFA), isoflavones, methionine and choline, and eicosapentaenoic acid (EPA) supplementation could reverse liver disease parameters [57–62]. In a double-blind placebo control, trial participants were supplemented with 460 mg/day of EPA and 380 mg/day of docosahexaenoic acid (DHA) for 15-18 months. In this study, subjects who were PNPLA3 148M/M carriers were associated with lower DHA tissue enrichment and hepatic fat levels. In contrast, EPA enrichment resulted in lower fasting triglyceride levels among 148M/M carriers [63]. In addition, a 4-month caloric restriction without changes in physical activity was performed and resulted in improvements in hepatic steatosis, anthropometric traits, and VLDL levels among subjects with the PNPLA3 and the transmembrane 6 superfamily 2 (TM6SF2) risk genotypes [64]. In contrast, in another study, patients with PNPLA3 148M risk variant had no changes in transient elastography with a controlled attenuation parameter (CAP) or liver enzymes after receiving 1840 mg EPA and 1,520 mg DHA for four weeks [65].
Furthermore, patients with the risk allele of rs738409 PNPLA3 had a low reduction in transaminase levels after supplementing with silymarin + vitamin E for six months [66]. Furthermore, in a small sample size clinical study, subjects were placed on a three-week hypercaloric rich in simple carbohydrates that induced DNL and liver fat accumulation. However, this effect was reversed after a six-month weight-loss period. In this study, there was a lack of a positive correlation between changes in DNL and triglycerides levels in PNPLA3-148MM carriers with defective triglyceride hydrolysis [67]. In a more recent study, the bodyweight loss resulted in liver stiffness changes among PNPLA3 CG/GG and HSD17B13 AG/GG carriers [68].
The lysophospholipid acyltransferase 7 (MBOAT7) is an enzyme that regulates phospholipid acyl-chain remodeling. The T-risk allele from MBOAT7 rs641738 reduces its protein expression, and as a consequence, there is a change in plasmatic phosphatidylinositol [69]. It is also associated with hepatic triglyceride accumulation and disease severity, including steatosis, inflammation, and fibrosis [70]. However, other studies have not found evidence of the association between rs641738 and NAFLD, NASH, fibrosis, or liver disease severity [71,72].
The Fat mass and Obesity-associated protein (FTO) is a demethylase enzyme [73]. It can down-regulate N6-methyladenosine, which is involved with increased triglyceride deposition [74]. One recent study found that FTO protein levels and gene expression were higher in samples from NAFLD and NASH patients during high-fat diet conditions. Due to this metabolic state, FTO seems to promote triglyceride accumulation and steatosis in the liver. This effect is possible because FTO blocks the PPARα transcription factor that would favor FAO [75]. Furthermore, FTO also regulates adipokines such as IL-6. A recent study has shown that FTO knockout mice fed with a high-fat diet increase the expression of IL-6 [76]. Different genetic variations (rs1421085 C-allele, rs8050136 A-allele, rs3751812 T-allele, and rs9939609 T-allele) in the FTO gene have been studied for their possible contribution role to the risk of NAFLD [77].
The homeostasis of biliary acid is important for the regulation of liver inflammation. One key gene polymorphism is rs17618244 G>A from the b-Klotho (KLB). KLB is a coreceptor of fibroblast growth factor receptor-4 (FGFR4) which interacts with FGF19 and induces the downregulation of cholesterol 7-alpha-hydroxylase (CYP7A1) and inhibits biliary acid synthesis. The rs17618244 minor A allele increases the risk of ballooning and lobular inflammation in children with NAFLD [78]. Another recent study found that the rs17618244 A allele was associated with hepatic fibrosis, lobular inflammation, and cirrhosis in patients with NAFLD. Moreover, its gene expression was higher in hepatic stellate cells, favoring the activity of profibrogenic and proliferative genes [79].
The enzyme carboxylesterase (CES1) detoxifies xenobiotics and activates ester and amide prodrugs. It also regulates the efflux of free cholesterol from macrophages to cholesterol acceptors and releases free cholesterol from lipoproteins in the liver for bile acid synthesis or direct secretion into the bile [80,81]. The genetic variation rc.428G>A (p.Gly143Glu) positively correlates with increased lipid storage and plasma concentration [82]. In addition, the deletion copy number variation (CNV) of CES1 on the 16q12.2 locus, in which carriers of < 2 CNV presented an increased risk of NAFLD [83].
5Nutrient-based prevention and treatment strategies5.1Dietary interventionsThe treatment of liver disease currently focuses on reducing hepatic lipid uptake while promoting weight loss. However, there are no precise medications or dietary guidelines to achieve this outcome. However, energy restriction is, until now, the most used strategy. For example, in a randomized control trial that examined the effect of a hypocaloric low carbohydrate diet (LCD) or a high carbohydrate diet (HCD), reductions in intrahepatic triglycerides were similar between groups [84]. Furthermore, in a randomized control trial, overweight and obese patients with type 2 diabetes followed a Paleolithic diet ad libitum combined with exercise for 12 weeks. At the end of the study, participants decreased fat liver content and improved peripheral and adipose tissue insulin sensitivity, but not hepatic insulin sensitivity [85]. In contrast, lifestyle changes consisting of balanced dietary modification and regular physical exercise reduced weight, hepatic lipid content, liver enzymes, and inflammatory markers [86,87].
As reported, DNL is key for liver disease progression; in this sense, a promising dietary strategy consisting of 4 g/day of EPA+DHA supplementation for 15-18 months was tested in a randomized control trial. The study found that participants who received ⩾2% of DHA improved hepatic insulin sensitivity, reduced fasting and postprandial plasma triglyceride levels, and decreased fasting hepatic DNL [88]. In another similar study, 4 g/day of EPA+DHA supplement for 8 weeks reduced fasting plasma triglycerides, intrahepatic triglycerides, fasting and postprandial hepatic DNL, and increased fatty acid oxidation in healthy participants [89].
5.2Antioxidants and vitaminsDifferent natural products have been used to exert antioxidant properties. In the case of liver disease, clinical studies with D-pinitol from carob pods helped to reduce fat liver accumulation and improved transaminases and triglycerides while reducing oxidative stress and inducing fatty oxidation [90]. In an experimental model, the administration of a white tea extract, an antioxidant compound, reversed steatosis by efficiently regulating the expression of lipid metabolism, oxidative stress, and inflammatory genes [91]. In another experimental model, C57BL/6 mice were fed with fructose to induce NAFLD and then were given vitamin E (70 mg/kg) for 2 weeks. As a result, vitamin E ameliorated glucose intolerance, liver steatosis, and lipid accumulation. These beneficial results were related to the upregulation of nuclear factor erythroid-2-related factor 2 (Nrf2), and CES1 downregulated lipogenic genes [92]. Vitamin E supplementation (400 mg twice a day) in patients with NASH helped decrease liver aminotransferases, IL-6, therefore showing an anti-inflammatory benefit [93]. Furthermore, the administration of a combined nutraceutical containing Vitamin D3, Vitamin E, Olive dry extract (Olea Europaea), Cinnamon dry extract (Cinnamomum cassia Presi cortex), and Fish oil 56% DHA/EPA was able to decrease lipid accumulation in the liver, mainly by increasing the expression of fatty acid oxidation enzymes, reducing lipoperoxidation products such as malondialdehyde (MDA)- and 4-hydroxy-2-nonenal, restoring complex I and by increasing the production of uncoupling protein 2 (UCP-2) [94]. The supplementation of plant extracts, L-Methionine, and L-Glutathione alone or in conjunction with a hypocaloric Mediterranean diet for six months helps to reduce weight, lipid profile, hepatic fat accumulation, and liver stiffness [95].
5.3Probiotics and prebioticsRestoration of gut microbiota is a promising strategy to treat and prevent many chronic diseases. As explained earlier, fructose induces microbiota dysbiosis associated with liver disease progression. However, this can be reversed when administered probiotics. For example, investigators studied the effect of the administration of Lactobacillus paracasei in C57BL/6 mice with NASH fed with 10% fructose and found remarkable results in reduced hepatic fat deposition serum ALT level, lower expression of proinflammatory genes [96]. Gut microbiota also can be manipulated with prebiotics and symbiotics. In line with this, supplementing with a symbiotic capsule with 200 million colony-forming units of 7 different bacteria strains, 125 mg fructo-oligosaccharide, magnesium stearate, and hydroxypropylmethylcellulose, there was a reduction in hepatic steatosis and fibrosis measured by transient elastography [97]. Probiotics also seem to improve liver histology and inflammatory markers in patients with NAFLD [98]. Ongoing randomized controlled trials aim to reduce fat liver content, inflammation, and fibrosis which are worthy of follow-up [99,100].
In summary, the growing evidence of nutrient-based strategies points out that they provide beneficial effects to counteract the nocive cycle of DNL, inflammation, and fibrosis in the context of obesity/NAFLD/NASH. Further studies are warranted to combine these strategies with a genome-based nutrition approach to optimize the effect of these interventions based on the genetic composition of the population [101].
6ConclusionIn the context of excess adipose tissue, as in obesity, metabolic syndrome, and T2DM, the activation of different molecular mechanisms such as insulin resistance, metabolic alteration, lipidic accumulation, oxidative stress, and inflammation increase the risk of developing liver disease. Additionally, altered dietary behaviors and unhealthy lifestyles favor dysbiosis that enhances unfavorable NAFLD-related mechanisms. Therefore, dietetic research that considers nutrient-based strategies to prevent and treat NAFLD is necessary.
CRediT authorship contribution statementIngrid Rivera-Iñiguez: Conceptualization, Writing – original draft, Writing – review & editing. Arturo Panduro: Conceptualization, Writing – original draft, Writing – review & editing. Sonia Roman: Writing – review & editing. Karina González-Aldaco: Writing – review & editing.
The research received no specific grant from funding agencies in the public, commercial, or not-for-profit sectors. Fundación Clínica Médica Sur provided the resources for the publication of this research.



