



Hepatocellular carcinoma (HCC) is one of the deadliest cancers. For patients with advanced HCC, liver function decompensation often occurs, which leads to poor tolerance to chemotherapies and other aggressive treatments. Therefore, it remains critical to develop effective therapeutic strategies for HCC. Etiological factors for HCC are complex and multifaceted, including hepatitis virus infection, alcohol, drug abuse, chronic metabolic abnormalities, and others. Thus, HCC has been categorized as a “genomically unstable” cancer due to the typical manifestation of chromosome breakage and aneuploidy, and oxidative DNA damage. In recent years, immunotherapy has provided a new option for cancer treatments, and the degree of genomic instability positively correlates with immunotherapy efficacies. This article reviews the endogenous and exogenous causes that affect the genomic stability of liver cells; it also updates the current biomarkers and their detection methods for genomic instabilities and relevant applications in cancer immunotherapies. Including genomic instability biomarkers in consideration of cancer treatment options shall increase the patients’ well-being.
Liver cancer is among the high-mortality cancers worldwide [1]. In some regions where infection of the oncogenic hepatic viruses is prevalent, liver cancer is the highest death-causing malignancy. Liver cancer is closely related to tumor virus infection and toxin metabolism. The hepatitis B and C virus infections induce chronic inflammation, conferring cytotoxic and genotoxic effects in the hepatocyte [2]. Expression of the oncogenic viral factors and their unique carcinogenic mutant proteins that emerge during the long-term chronic infection periods results in gain-of-growth and cell transformation. Moreover, toxic products of endogenous and environmental metabolites accumulate in the liver, increasing the liver burden in genomic instability. Hepatocellular carcinoma (HCC), the most common type of liver cancer, was generally high in genomic instability [3]. The hepatocyte often responds to cellular stress by skipping cell division, which leads to polyploidy and subsequent aneuploidy [4]. According to various analyses, such as whole-genome sequencing, global gene mutation rates, chromosome aberrations, and gene copy number variations, HCC harbors higher genomic instability than many other solid tumors, even in the early stage of malignant change [5]. These findings have suggested that hepatocyte genomic instability plays a vital role in the initiation and progression of cancer. Nevertheless, the status of the genomic instability in the tumor has also been identified as an important predictive biomarker for susceptibilities to chemotherapies and immune therapies. Molecular biomarkers such as microsatellite instability (MSI) and tumor mutation burden (TMB) have been widely examined to predict therapeutic efficacies [6]. Thus, developing new genomic stability-relevant biomarkers that can be used as valuable tools for evaluating the tumor's susceptibilities to new anticancer drugs is critical. This article will review the leading causes of hepatocyte genomic instabilities for their effects on liver pathogenesis and malignancy. We will also update the recent developments of genomic instability biomarkers for predicting the therapeutic efficacies for various anticancer treatments. Perspectives on developing new biomarkers in the relevant fields will also be discussed.
2Hepatitis B virus infection-induced genomic instabilityHBV is a lipid-enveloped hepatotropic virus and has a partially double-stranded relaxed circular DNA (rcDNA) genome, which is enclosed within a protein capsid and contains four overlapping open reading frames (ORFs; P, S, C, and X) [2]. Studies have found that HBV infection induces genomic instability. The HCC with HBV infection presents with higher genomic instability than no HBV infection [7]. Several mechanisms have been proposed to elucidate the development of HBV-related HCC, including the chronic inflammation initiated by host immune responses to viral infection, the insertional mutagenesis caused by viral DNA integration into host cell genomes, and the oncogenic functions executed by viral proteins [8-10]. Moreover, these mechanisms have also been implicated in the induction of genomic instability in chronic HBV infection, contributing to HCC development [11]. Indeed, genomic instability is frequent in the multistep process of chronic HBV infection-associated hepatocarcinogenesis.
2.1HBV proteins-induced genomic instability2.1.1HBV X proteinHBV X protein (HBx) is small, 154 amino acids long, nonstructural, and important in the hepato-carcinogenic processes. HBx is multifunctional, activates cellular signaling pathways, and is essential for viral infection [12]. It interacts with nuclear transcription factors and modulation of cytoplasmic signal transduction pathways, including the Ras, Raf, c-jun, MAPK, NFkB, Jak-Stat, FAK, and protein kinase C pathways, as well as Src-dependent and phosphatidylinositol-3 kinase signaling cascades, leading to hepatocellular carcinogenesis [13]. Furthermore, HBx binds to the carboxy (COOH)-terminus of the p53 protein and inhibits its binding to the nucleotide excision repair proteins xeroderma pigmentosum B (XPB) and XPD [14]. HBx also interacts with the damaged DNA binding (DDB) protein 1-containing E3 ubiquitin ligase to target the structural maintenance of chromosomes (SMC) complex SMC5/6 for degradation, leading to abrogation of DNA repair and chromatin organization [15]. In addition, through its negative effect on p53, HBx contributes to anti-p53-mediated apoptosis and promotes cell proliferation [16]. Taken together, attributed to its impact on inhibiting functions of several DNA repair factors and apoptosis, HBx plays a significant role in causing genomic instability in the host cell.
2.1.2HBV surface protein and the pre-S variantsThe HBV surface (HBS) protein is the main component comprising the viral envelope. The HBS gene is transcribed into the pre-S1 and pre-S2/S transcripts, which differ in the transcriptional start sites but share the same termination sites. These transcripts are translated into three different surface proteins that start from the pre-S1, pre-S2, and S regions, leading to the production of the large, medium, and small surface proteins, respectively [17]. The pre-S1 region on the large surface protein contains the binding site to the HBV receptor Na+-taurocholate cotransporting polypeptide (NTCP) on the hepatocyte surface, therefore it is essential for viral entry [18]. Partial deletions in the pre-S1 and pre-S2 regions have been found to be highly prevalent in the serum and liver of the HBV-related HCC [19,20]. The pre-S mutants are the mutant forms of large HBS (LHBS) protein which harbors in-frame deletion mutations over the pre-S1 or pre-S2 domains. Due to the deletion-induced protein misfolding, these pre-S mutants predominantly accumulate in the endoplasmic reticulum (ER), and trigger unfolded protein response (UPR) signaling pathways [17,21]. Intracellular accumulation of the misfolded mutant LHBS protein results in the histological morphology of ground glass hepatocyte (GGH), characterized by unclear/cloudy pattern in the cytoplasm upon HE staining [22]. Pathological observations have found that GGH is prevalent in HBV-related HCC tissues, suggesting that the pre-S mutant LHBS is associated with the carcinogenic process of the liver [23-25]. Further studies in our group also found that the pre-S mutant LHBS induces ER stress-mediated oxidative stress, DNA damage, and gene mutations [17,26-29]. Moreover, we found that the pre-S2 deletion mutation reveals a new conformation in the pre-S region and results in a new virus-host interaction [29]. The pre-S2 mutant LHBS interacts with C-Jun activation domain-binding protein 1 (JAB1), causing degradation of the cyclin-dependent kinase inhibitor p27Kip1 and a failure in the cell cycle checkpoint [29]. Therefore in the pre-S2 mutant LHBS(+) cells, continuous cell cycle progression in the presence of DNA damages leads to significant genomic instability, a pre-requisite for cancer. Furthermore, studies in the HBV transgenic mice found that, compared with HBx transgenic mice, the pre-S2 mutant LHBS mice showed significantly higher gene copy number variations (CNVs) as the biomarker for global genomic instability [28]. Interestingly, the pre-S2 mutant LHBS was also found to specifically interact with importin α1 and consequently impairs its nuclear transport function [28]. This effect caused a nuclear translocation deficiency of DNA double-strand break (DSB) repair factor Nijmegen breakage syndrome 1 (NBS1), a key regulator of the MRN complex essential for the sensing and early processing of DNA DSB [28]. Taken together, the pre-S2 mutant LHBS, prevalent in chronic HBV (CHB), represents a major viral oncoprotein conferring genomic instability in the carcinogenic process.
2.1.3HBV splicing variantsStudies have shown that the HBV RNA can be alternatively spliced to generate at least 14 splice variants that have been identified in sera and livers of chronic hepatitis B (CHB) patients [30]. It has been reported that up to 80% of intracellular capsids contain the viral DNAs originating from the spliced RNAs in the HBV genome-replicating hepatoma cells [31]. Although RNA splicing of hepatitis B virus (HBV) is commonly observed in the livers of hepatitis B patients and in the cultured cells replicating the viral genome, its biological significance in the HBV life cycle and the detailed regulatory mechanisms are still largely unclear. Studies have shown that some splicing variants from pre-genomic (pg) and pre-S2/S RNA are associated with inflammation, cirrhosis, and carcinogenic processes [30,32]. The major spliced variant termed SP1, which has an intron between nt 2448 and 488, may account for up to 30% of pgRNA but vary in different viral genotypes [33]. Though approximately one-third of the viral genome is deleted, the splice variant SP1 RNA has been shown packaged to generate defective HBV circulating particles [34]. Soussan and his colleagues found that in the mice with the humanized liver, treatments with genotoxins resulted in liver damage and a significant increase of HBV splice variants, implicating that DNA damage induces the generation of viral splicing variants [35]. Furthermore, studies by Chen et al. [36] also found that increased proportions of HBV splice variants were associated with the resistance of CHB patients to interferon therapies and the severities of liver diseases, including HCC. The SP1 RNA translational product, hepatitis B spliced protein (HBSP), interacts with and activates the microsomal epoxide hydrolase (MEH) enzyme, which transforms the DNA-damaging chemical benzo[a]pyrene into a potent carcinogen [37]. Taha et al. reported that HBV pgRNA splicing event is modulated and stabilized by histone deacetylase 5 (HDAC5), suggesting that developing specific inhibitors to block the viral RNA-HDAC5 association is likely a potentially novel approach in antiviral therapies [38].
2.2HBV viral integrationIntegration of HBV DNA into the human genome is prevalent and occurs within several days after viral entry [39,40]. It is considered an early event during hepatocarcinogenesis. A report by Zhao et al. indicated that HBV integration occurs in 1 of approximately 105-106 infected hepatocytes since the early phases of infection and in up to 90% of tumor cells from patients with HBV-related HCC [41]. HBV integration can induce chromosomal instability [42]. Integration of HBV DNA into the host genome causes genetic damage and insertional mutagenesis of the genes involved in carcinogenesis [9]. Many integration hotspots have been identified near or inside cancer-related genes, such as telomerase reverse transcriptase (TERT), cyclin A2 (CCNA2), and p53, leading to enhanced proliferation and transformation [43]. Studies by Zhao et al. reported that HBV integration sites were significantly enriched in the proximity of telomere in the tumor samples, leading to telomere dysfunction, extensive DNA amplification, and large terminal deletions [44]. Some reports also showed that HBV DNA preferentially integrates into fragile sites such as intergenic regions, repetitive regions, CpG islands, and genome telomeres [39]. This event leads to genomic instability and abnormal gene expression in the affected chromosomal areas. The rates of viral integration in the infected cells are greatly affected by the environmental or endogenous genotoxin-generating DNA DSB, which offers a preferential integration site for viral-host gene fusion, evidenced in both the duck and human HBV infection cell models that have reported similar findings [45]. Furthermore, it was reported that approximately 10% of the HBV-infected cells present with viral double-stranded linear DNA (dslDNA) [39]. The viral dslDNA, given its open DSB ends, is potent in integrating into the host chromosome. Some studies also proposed that HBV DNA integration is likely carried out by several DSB repair pathways, such as the sequence-independent non-homologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ) pathways, indicating that DNA DSB is a major triggering factor for viral integration [46].
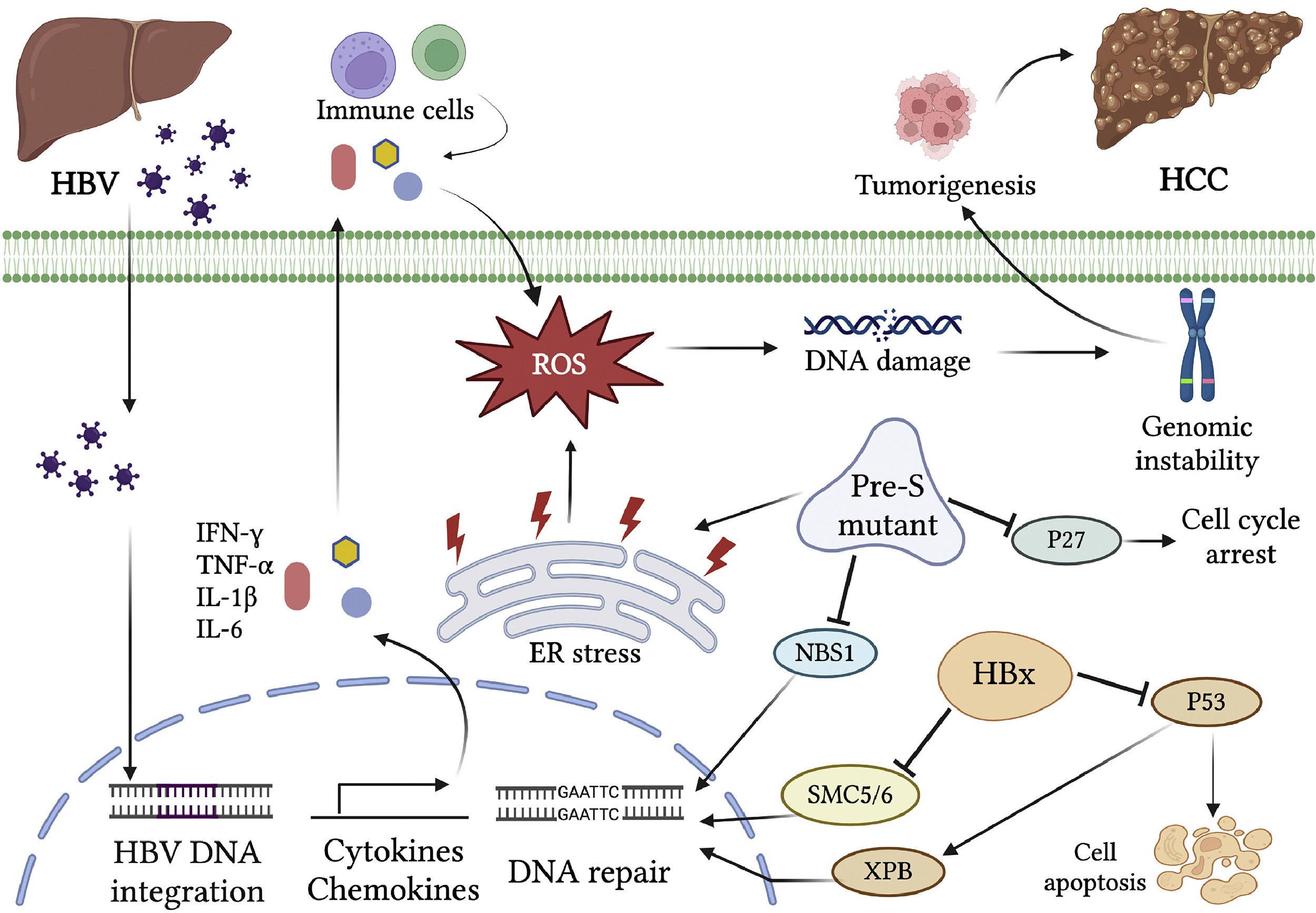
2.3HBV chronic inflammation-induced genomic instabilityThe hepatitis virus infection causes the activation of inflammation responses and secretion of various cytokines and chemokines. The pro-inflammatory cytokines such as interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) can activate the intracellular signals for reactive oxygen species (ROS) production through affecting mitochondrial NADPH oxidase activities [47,48]. They also recruit immune cells to the sites of infection, where further induction of inflammatory cytokines is triggered. In addition, numerous inflammatory cytokines such as IL-1β, IL-6, IL-15, and TNF-α, and chemokines such as IL-8 and GRO-alpha, induce the production of ROS and reactive nitrogen species (RNS), both resulting in DNA damage [49]. The immune clearance phase of HBV infection is characterized by increased activation of host immune responses. The immune cells, mainly natural killer cells, and HBV-specific cytotoxic T cells are activated to recognize and destroy the virus-infected hepatocytes [50]. Nevertheless, Cho et al. found that excessively activated CD8 (+) T cells induce enormous inflammatory responses in this phase. In some cases, prolonged immune reactions due to failure of complete viral clearance by immune cells in this phase result in chronic inflammation, liver injury, and increased risks of liver diseases such as cirrhosis and HCC [51]. The immune response-induced oxidative stress damages cellular DNA, proteins, or lipids and consequently affect cellular signaling pathways and gene expression profiles related to cell cycle, apoptosis, growth, adhesion, and metabolism [52]. Indeed, elevated levels of oxidative stress and lipid peroxidation in CHB patients were found to be positively correlated with serum viral titers [53]. Cervantes-Gracia and his colleagues have also reported that inflammatory reactions-induced ROS/RNS in CHB lead to the aberrant expression of a DNA mutator enzyme, the activation‐induced cytidine deaminase (AID), which causes somatic mutations and epigenetic alterations that result in inhibition of p53 and reactivation of TERT expression [54]. In fact, AID activation is widely observed in gastrointestinal tissues with cancer‐associated inflammation and chronic viral hepatitis [55,56]. Taken together, the chronic inflammation responses in CHB play a critical role in HBV-related genomic instability and tumorigenesis (Figure 1).

Genomic instability caused by chronic HBV infection. HBV infection triggers the activation of inflammation responses and secretion of various cytokines and chemokines. The inflammatory cytokines, such as IL-1β, IL-6, IL-15, and TNF-α, and chemokines, such as IL-8 and GRO-alpha, lead to the production of ROS and RNS, both resulting in DNA damage. In addition, the viral proteins interact with host factors and activate the carcinogenic pathways. HBx binds to p53 and inhibits its function in apoptosis and DNA repair. HBx also targets the SMC5/6 complex for degradation, leading to the abrogation of DNA repair. The HBV surface protein pre-S mutant predominantly accumulates in the ER and activates the ER stress signaling pathway, which induces oxidative DNA damage and gene mutation. The pre-S2 mutant LHBS causes degradation of the cyclin-dependent kinase inhibitor p27Kip1 and a failure in the cell cycle checkpoint. It also causes a nuclear translocation deficiency of DNA DSB repair factor NBS1, leading to genomic instability. Integration of HBV DNA into the host genome causes genetic damage and insertional mutagenesis of the genes involved in carcinogenesis. Chronic HBV infection induces genomic instability through multiple processes, which render the hepatocyte at high risk of hepatocarcinogenesis.
Hepatitis C virus (HCV) infection is associated with the development of HCC and non-Hodgkin's B cell lymphoma. HCV infection induces chromosome aberrations through inhibiting multiple DNA repair processes, leading to genomic instability in monocytes and hepatocytes [57]. It was found that liver cell lines constitutively expressing the HCV genome show a high incidence of chromosome instability, aneuploidy, and mitotic impairments [58]. HCV inhibits ROS-induced DNA damage repair and interferes with the ataxia-mutated (ATM)/Nijmegen breakage syndrome protein 1 (NBS1)/Mre11/Rad50 DNA repair pathway for double-strand break (DSB) [59,60]. Previous studies reported that HCV induced the expression of the error-prone DNA polymerase ζ, polymerase ι, and activation-induced cytidine deaminase (AID), contributing to a mutator phenotype [61,62]. Furthermore, HCV infection causes ROS-mediated metabolic dysfunctions, including lipid peroxidation and steatosis [63,64]. Some HCV proteins, such as structural nucleocapsid protein core and nonstructural viral protein NS3 and NS5A, have been found to directly interact with and inhibit proteins involved in repairing DSB and oxidative DNA damage [65]. These effects together contribute to genomic instability that promotes the carcinogenic process. Nevertheless, the HCV-induced mutator phenotype can be a potential target for cancer immunotherapy with immune checkpoint inhibitors such as the anti-PD-1/PD-L1 antibody drugs [66]. The effects of genomic instability attributed to the specific HCV factors are summarized below.
3.1HCV proteins-induced DNA repair inhibition3.1.1HCV core proteinThe HCV core protein is the key structural protein in the viral capsid, a spherical structure that surrounds and directly binds to the viral genomic RNA [67]. In the HCV-infected cells, some core proteins translocate to mitochondria and inhibits the electron transport chain (ETC) reaction, accumulating the oxidative adducts nitric oxide (NO) and ROS [68]. It leads to chronic oxidative stress causing chromosomal and mitochondrial DNA instability and lipid peroxidation in the infected cell [69]. Additionally, it binds to the DNA DSB repair factor NBS1 and blocks the formation of the Mre11/NBS1/Rad50 complex for DNA repair [70]. It was also found that the HCV core protein binds to and inhibits activation of the DNA damage sensor protein ATM, leading to impaired DNA DSB repair [57]. Furthermore, the core protein was found to reduce the DNA glycosylase activity in repairing and excising the oxidative adduct 8-oxoguanine in the genome. This reaction is likely mediated through the interaction of the core protein with the c-Jun oncogenic transcription factor [70].
3.1.2HCV nonstructural protein 3Nonstructural protein 3 (NS3) is the main HCV serine protease responsible for the cleavages of the viral polyprotein and maturation of the nonstructural proteins [71]. NS3 also proteolytically inactivates host innate immune factors and escapes the virus from anti-viral immune surveillance. Studies have shown that HCV NS3 protein enhances matrix metallopeptidase 9 (MMP9) and COX-2 levels, triggering oncogenic signaling pathways for HCC [72]. Moreover, a previous study found that the NS3 protein interacts with and inhibits DNA repair factors Werner syndrome protein (WRN) and Ku70, impairing the WRN-mediated DNA repair and nonhomologous end joining (NHEJ) [73]. Additionally, the NS3/4A protease cleaves WRN and promotes its degradation by the 26S proteasome, reducing the WRN-mediated DNA repair [73]. Through this, NS3/4A impairs DNA repair and renders the infected cell to an accumulation of DNA mutations and genome instability.
3.1.3HCV nonstructural protein 5ANonstructural protein 5A (NS5A) is a zinc-binding and proline-rich hydrophilic phosphoprotein that plays a crucial role in HCV RNA replication, subcellular localization, and egress [74]. NS5A is derived from a large polyprotein translated from the HCV RNA genome and undergoes post-translation processing by the NS3 viral protease [75]. NS5A has been reported to interact with some viral and host proteins in the infected cell. NS5A binds to and facilitates the polymerase activity of the viral RNA-dependent RNA polymerase (RdRp) NS5B for viral replication [76]. The overexpression of HCV NS5A protein in cultured liver cells promoted chromosome instability and aneuploidy [58]. A previous study showed that HCV NS5A directly bound the DNA DSB repair factor RAD51-associated protein 1 (RAD51AP1) and impaired DNA repair function of the RAD51/RAD51AP1/UAF1 complex, leading to chromosome instability and mitotic impairments of the infected cells [77]. The HCV NS5A inhibits DNA DSB repair and induces genomic instability, a crucial molecular mechanism of HCV-associated hepatocarcinogenesis.
In summary, HCV proteins inhibit the activities of multiple DNA repair pathways through interaction with host DNA repair factors. The infection also induces error-prone DNA polymerases and increases mutation frequencies. These interplays between the virus and its host in the infected cell are crucial in HCV-associated carcinogenesis. Thus, the genomic instability phenotype caused by HCV infection provides a promising target for cancer immunotherapies using relevant immune checkpoint inhibitors.
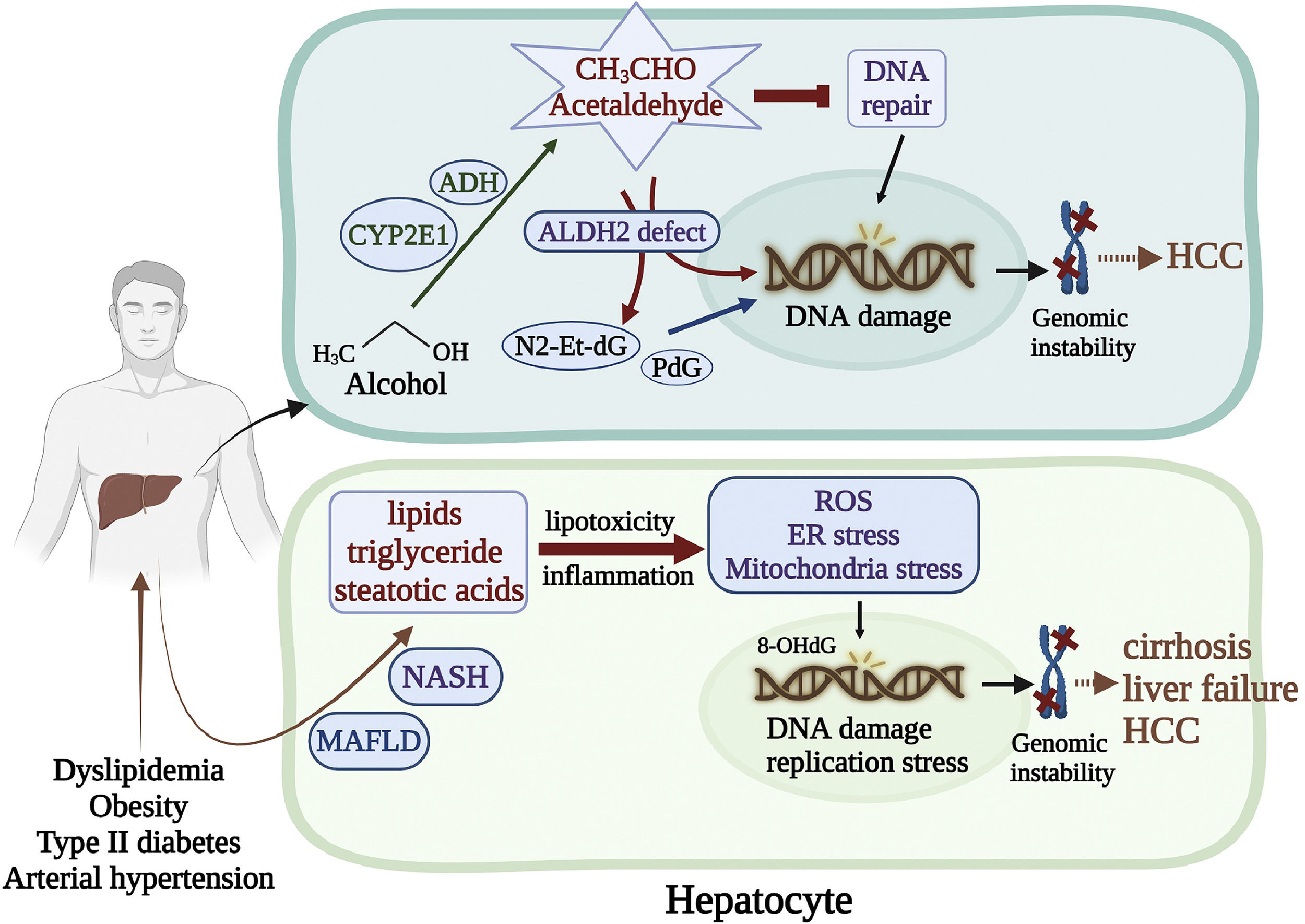
4Genomic instability caused by alcohol and nonalcoholic metabolic liver diseases4.1Alcohol-related genomic instabilityStudies have shown that alcohol causes DNA damage and cancer in genetically modified mice. The alcohol breakdown product acetaldehyde is a potent genotoxin. Acetaldehyde is a highly reactive compound that can cause various DNA damage, such as base substitutions, DNA-DNA, and DNA-protein crosslinks [78,79]. In addition, acetaldehyde interacts with some DNA repair proteins and inhibits their functions [80]. Acetaldehyde also induces deoxynucleotide adducts such as N2-ethyl-2′-deoxyguanosine (N2-Et-dG) and N2-propano-2′-deoxyguanosine (PdG), which are highly mutagenic [81]. A recent study by Guidolin et al. showed that the DNA adducts N6-ethyl deoxyadenosine and N4-ethyl deoxycytidine were detected in human oral DNA 2 hours after alcohol exposure [82]. The most important natural defense enzyme for acetaldehyde toxicity is aldehyde dehydrogenase 2 (ALDH2) [83]. A study by Weng et al. revealed that DNA damage was exacerbated in the ALDH2 knock-out (KO) mice [84]. It is worth noting that ALDH2 deficiency (rs671, ALDH2*2, also known as the flushing mutation) affects approximately 35%-40% of East Asians, and 8% of the world population, who are in increased risks of acetaldehyde-associated cytotoxic and genotoxic effects [85]. In addition, it was reported that the ALDH2 rs671 polymorphism was associated with increased risks to HCC and esophageal cancer and HBV/HCV-related cirrhosis with mild alcohol intake [86]. Moreover, other studies have indicated variations of the alcohol-related metabolic enzymes, such as alcohol dehydrogenase (ADH) and cytochrome P450 2E1 (CYP2E1), which regulate the conversion of alcohol into acetaldehyde, are associated with DNA damage in the hepatocyte [87]. Likely, decreased expressions of the CYP2E1 and ADH genes were susceptible to HCC and esophageal cancers in alcohol consumers and HBV/HCV infection [88,89].
4.2Metabolic-associated fatty liver disease-induced genomic instabilityMetabolic-associated fatty liver disease (MAFLD) has become the most common liver disorder worldwide. It is present in approximately 25% of the world's population, common in developed nations such as the United States, where about 100 million individuals were affected in 2017 [90]. MAFLD is related to dyslipidemia, obesity, type 2 diabetes, and arterial hypertension [91]. Some individuals with MAFLD can develop nonalcoholic steatohepatitis (NASH), marked by fatty liver-associated inflammation, cirrhosis, and potentially liver failure. Accumulation of excessive lipids such as triglyceride, free steatotic acids, and their metabolites in the liver causes lipotoxicity, triggering a cascade of stress-induced responses in the organelles ER and mitochondria and ROS generation [92,93]. In mouse models of NASH, the dividing steatotic hepatocytes exhibited oxidative stress and activation of the DNA damage response (DDR) [94]. This effect is considered a crucial determinant of cancer development associated with MAFLD. Liver biopsies from MAFLD patients were found that the levels of the oxidized nucleotide adduct 8-OHdG were related to the stage of fibrosis and serum α-fetoprotein (AFP) levels [95]. Moreover, Donne et al. reported that livers from MAFLD patients displayed nucleotide pool imbalance and replication stress, leading to DNA damage, cGAS/STING pathway activation, and type I interferon expression [96] (Figure 2).
 The alcohol breakdown product acetaldehyde is a highly reactive compound that interacts with DNA repair proteins and inhibits their functions. Acetaldehyde also induces deoxynucleotide adducts, such as N2-Et-dG PdG, which are highly mutagenic. ALDH2 is the most critical defense enzyme against the acetaldehyde toxicity. Mutation of the ALDH2 gene renders the hepatocyte susceptible to HCC. (Bottom) MAFLD is related to dyslipidemia, obesity, type 2 diabetes, and arterial hypertension. Some individuals with NAFLD can develop NASH, marked by fatty liver-associated inflammation, cirrhosis, and potentially liver failure. Accumulation of excessive lipids in the liver triggers a cascade of stress-induced responses in the ER and mitochondria, leading to oxidative DNA damage and genomic instability.")
Genomic instability caused by alcohol and MAFLD. (Top) The alcohol breakdown product acetaldehyde is a highly reactive compound that interacts with DNA repair proteins and inhibits their functions. Acetaldehyde also induces deoxynucleotide adducts, such as N2-Et-dG PdG, which are highly mutagenic. ALDH2 is the most critical defense enzyme against the acetaldehyde toxicity. Mutation of the ALDH2 gene renders the hepatocyte susceptible to HCC. (Bottom) MAFLD is related to dyslipidemia, obesity, type 2 diabetes, and arterial hypertension. Some individuals with NAFLD can develop NASH, marked by fatty liver-associated inflammation, cirrhosis, and potentially liver failure. Accumulation of excessive lipids in the liver triggers a cascade of stress-induced responses in the ER and mitochondria, leading to oxidative DNA damage and genomic instability.
Genome instability is a vital factor in the occurrence and progression of cancer. Due to various genotoxic causes such as viral infection, environmental stimuli, and metabolic abnormalities, liver cancer generally shows a significant increase in gene mutations [97]. The accumulation of gene mutations leads to the instability of the genome, which even affects prognosis and the treatments of choice for cancer. Developing applicable biomarkers of genomic instability is critical for assessing cancer characteristics and treatment options. The characterization of genomic instability in liver cancer has been investigated in many studies, and the chromosomal instabilities, including translocation, deletions, and inversions, were reported [5,98]. We also found that HBV-infected hepatocytes showed higher oxidative DNA lesion 8-OHdG and gene mutation frequencies [26]. Therefore, the level of intracellular 8-OHdG is likely a promising biomarker representing oxidative stress and relative risk to cancer initiation due to the accumulation of gene mutations [26,27]. Moreover, recent studies have identified some genomic instability-relevant predictive biomarkers for the efficacies of cancer therapies. The two major biomarkers microsatellite instability (MSI) and tumor mutation burden (TMB), in relation to treatment responses, are described here:
5.1.1Microsatellite instabilityMicrosatellite instability is a phenomenon attributed to DNA deletions and insertions caused by the deficiency in DNA mismatch repair (MMR) [99]. Studies found that MSI has different and significant impacts on the efficacies of cancer treatments. MSI has been shown to be negatively correlated with the effectiveness of the DNA-damaging chemotherapy drug 5-fluorouracil (5-FU). Jo et al. reported that patients whose colorectal tumors were MSI-high (MSI-H) did not gain a survival benefit with 5-FU compared to patients with microsatellite stable (MSS) tumors [100]. In addition, the MSI-H colon cancer cell lines were more resistant to 5-FU than MSS cell lines. More specifically, the research group found that DNA MMR proteins recognized and bound to 5-FU-DNA adducts, which triggered G2/M cell cycle arrest and, consequently cell death [100].
On the contrary, genomic instability in the tumor enhances the killing effects of immunotherapies. The ones with high MSI responded better to the humanized programmed cell death-1 (PD-1) blocking antibodies than otherwise [101,102]. High genomic instability, represented as MSI-H, enhances neoantigen production and stimulation of T-cell immune surveillance, leading to favorable responses to PD-1/PD-L1 immune therapies [103]. In 2019, the European Society for Medical Oncology (ESMO) recommendations proposed using the MMR protein immunohistochemistry (IHC) and MSI-PCR tests to recognize cancers with hypermutability characteristics. The use of multiple poly-A mononucleotide MS markers was recommended for the MSI-PCR tests to reach high sensitivities and specificities [104]. However, the global MSI status in HCC remains to be characterized by comprehensive cohort studies. Thus, Chiappini et al. found that MSI-H (unstable MS markers > 30%) was identified in approximately 20% and MSI-low (MSI-L, unstable markers < 30%) in 27% of HCC patients in a cohort study. And MSI-H was significantly associated with more aggressive histological tumor features and a shorter median delay before recurrence [105]. Nowadays, MSI has been widely applied as an important predictive biomarker for treatment responses to the cancer PD-1/PD-L1 antibodies such as nivolumab or pembrolizumab [106].
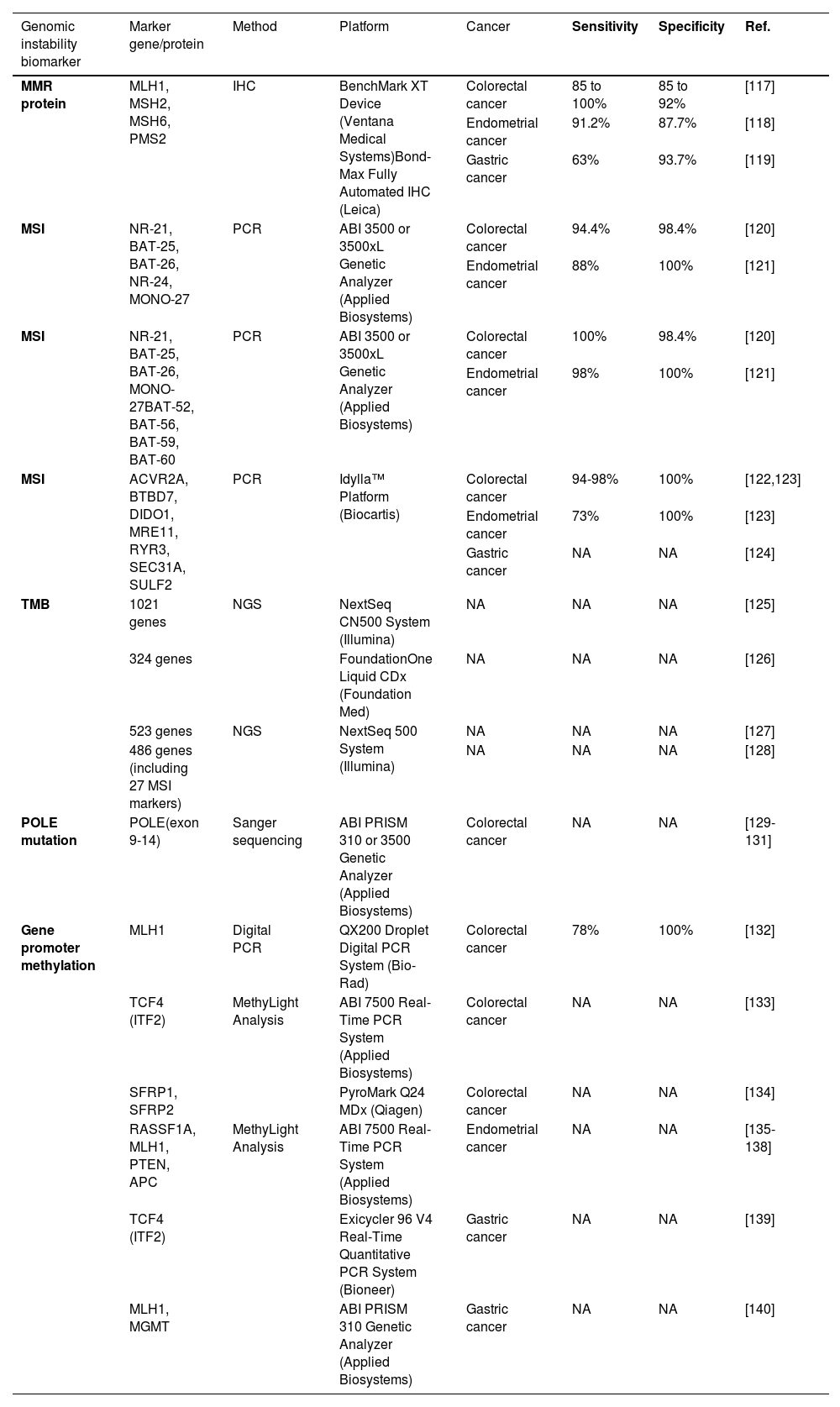
5.1.2Tumor mutation burdenLike MSI, tumor mutation burden (TMB), measured by whole-exome sequencing or multigene cancer panels, can predict therapeutic efficacies of immune checkpoint inhibitors (ICI) in multiple cancer types [107]. High mutation rates in somatic cells are believed to amplify the neoantigen load, leading to lymphocyte activation and enhanced cancer susceptibility to immunotherapies. One cross-cancer study by Cho et al. reported that a greater TMB was associated with higher PD-L1 expression in tumor cells and was positively correlated with responses to nivolumab or pembrolizumab [108]. Several studies have also shown that a higher TMB was associated with immune microenvironment diversification and worse prognosis in HCC patients [109,110]. Thus, the use of TMB as a significant predictive biomarker for immunotherapy efficacies in HCC remains to be extensively examined by cohort studies. The current genomic instability biomarkers and respective detection methods for the responses to cancer immunotherapies are summarized in Table 1.
Tests for genomic instability biomarkers in cancer immunotherapies
| Genomic instability biomarker | Marker gene/protein | Method | Platform | Cancer | Sensitivity | Specificity | Ref. |
|---|---|---|---|---|---|---|---|
| MMR protein | MLH1, MSH2, MSH6, PMS2 | IHC | BenchMark XT Device (Ventana Medical Systems)Bond-Max Fully Automated IHC (Leica) | Colorectal cancer | 85 to 100% | 85 to 92% | [117] |
| Endometrial cancer | 91.2% | 87.7% | [118] | ||||
| Gastric cancer | 63% | 93.7% | [119] | ||||
| MSI | NR-21, BAT-25, BAT-26, NR-24, MONO-27 | PCR | ABI 3500 or 3500xL Genetic Analyzer (Applied Biosystems) | Colorectal cancer | 94.4% | 98.4% | [120] |
| Endometrial cancer | 88% | 100% | [121] | ||||
| MSI | NR-21, BAT-25, BAT-26, MONO-27BAT-52, BAT-56, BAT-59, BAT-60 | PCR | ABI 3500 or 3500xL Genetic Analyzer (Applied Biosystems) | Colorectal cancer | 100% | 98.4% | [120] |
| Endometrial cancer | 98% | 100% | [121] | ||||
| MSI | ACVR2A, BTBD7, DIDO1, MRE11, RYR3, SEC31A, SULF2 | PCR | Idylla™ Platform (Biocartis) | Colorectal cancer | 94-98% | 100% | [122,123] |
| Endometrial cancer | 73% | 100% | [123] | ||||
| Gastric cancer | NA | NA | [124] | ||||
| TMB | 1021 genes | NGS | NextSeq CN500 System (Illumina) | NA | NA | NA | [125] |
| 324 genes | FoundationOne Liquid CDx (Foundation Med) | NA | NA | NA | [126] | ||
| 523 genes | NGS | NextSeq 500 System (Illumina) | NA | NA | NA | [127] | |
| 486 genes (including 27 MSI markers) | NA | NA | NA | [128] | |||
| POLE mutation | POLE(exon 9-14) | Sanger sequencing | ABI PRISM 310 or 3500 Genetic Analyzer (Applied Biosystems) | Colorectal cancer | NA | NA | [129-131] |
| Gene promoter methylation | MLH1 | Digital PCR | QX200 Droplet Digital PCR System (Bio-Rad) | Colorectal cancer | 78% | 100% | [132] |
| TCF4 (ITF2) | MethyLight Analysis | ABI 7500 Real-Time PCR System (Applied Biosystems) | Colorectal cancer | NA | NA | [133] | |
| SFRP1, SFRP2 | PyroMark Q24 MDx (Qiagen) | Colorectal cancer | NA | NA | [134] | ||
| RASSF1A, MLH1, PTEN, APC | MethyLight Analysis | ABI 7500 Real-Time PCR System (Applied Biosystems) | Endometrial cancer | NA | NA | [135-138] | |
| TCF4 (ITF2) | Exicycler 96 V4 Real-Time Quantitative PCR System (Bioneer) | Gastric cancer | NA | NA | [139] | ||
| MLH1, MGMT | ABI PRISM 310 Genetic Analyzer (Applied Biosystems) | Gastric cancer | NA | NA | [140] |
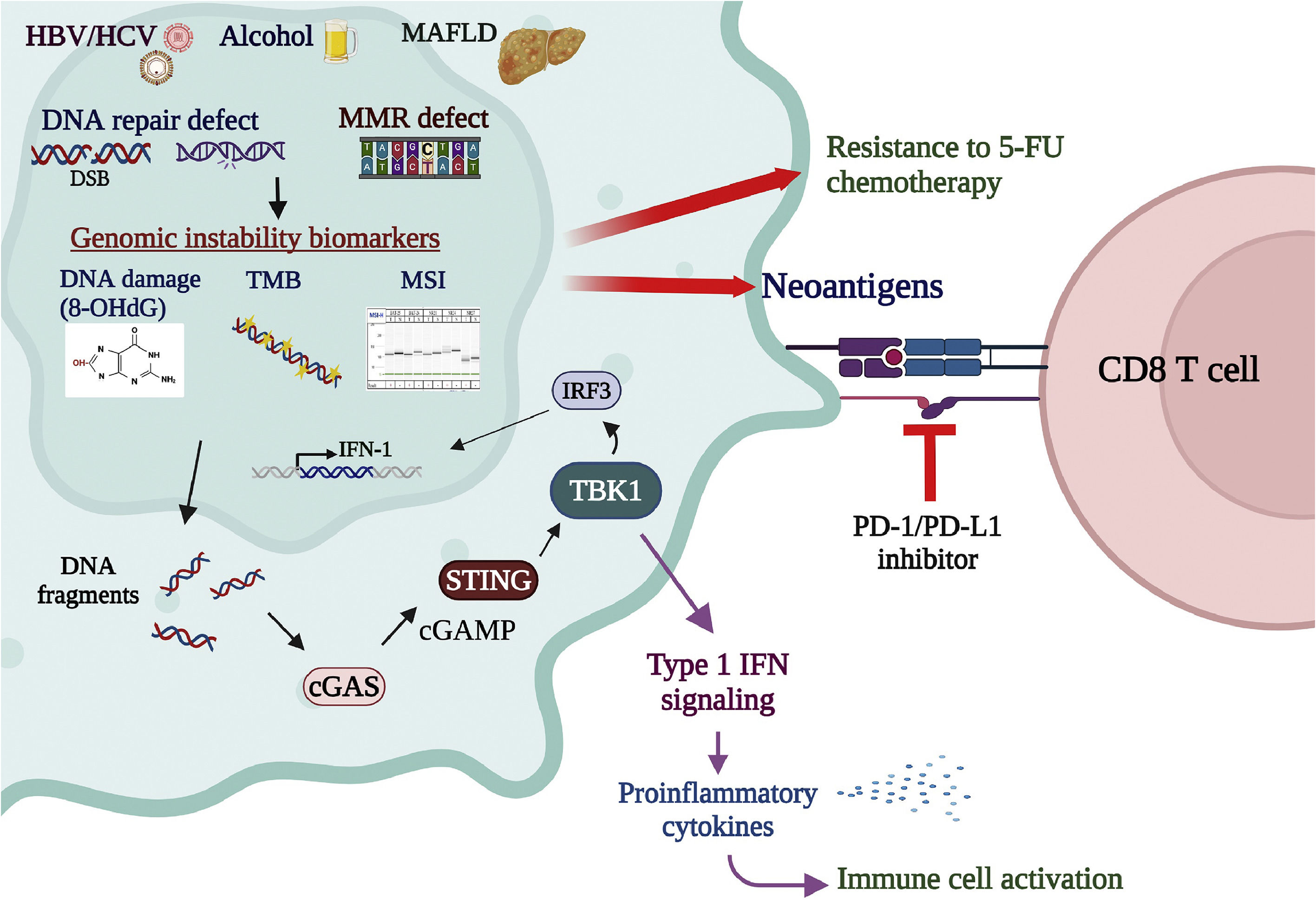
DNA fragmentation caused by DNA repair deficiency activates the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway, which senses cytosolic DNA fragmentation and activates the immune responses in the tumor microenvironment [111]. The cGAS-STING pathway plays a crucial role in activating the immune system and has been recently proposed as a promising target in cancer immunotherapies [112]. Activation of the cGAS-STING pathway co-administrated with other cancer immunotherapies, such as ICI inhibitors and antibodies, is thought to enhance the tumor-killing effects [113]. In addition, the STING downstream molecule TANK-binding kinase 1 (TBK1), which mediates the cGAS-STING signaling for type I interferon expression, was also shown to be a promising target for cancer immunotherapies [114]. In the tumor cell, the status of genomic instability, presented by various biomarker panels such as MSI, TMB, and cGAS-STING activation, is important for evaluating treatment responses to anticancer chemotherapies and immunotherapies [115,116]. Comprehensive cohort studies to explore the correlations among various genomic stability biomarkers and novel anticancer therapeutic approaches are upcoming critical tasks (Figure 3).

Genomic instability biomarkers associated with cancer therapies. MSI, representing deficiency in DNA MMR, is correlated with resistance to the 5-FU chemotherapy. On the contrary, genomic instability in the tumor enhances the killing effects of immunotherapies. MSI-H enhances neoantigen production and stimulation of T-cell immune surveillance, leading to favorable responses to PD-1/PD-L1 antibody immune therapies. TMB, measured by whole-exome sequencing or multigene cancer panels, is also a predictive biomarker for response to immune checkpoint inhibitors. High TMB amplifies the neoantigen load, activating lymphocytes and enhancing cancer susceptibility to immunotherapies. DNA fragmentation caused by DNA repair deficiency activates the cGAS-STING pathway, crucial in activating immune responses. Activation of the cGAS-STING pathway co-administrated with other cancer immunotherapies, such as ICI inhibitors and antibodies, is thought to enhance the tumor-killing effects. In addition, the STING downstream molecule TBK1, which mediates the cGAS-STING signaling for type I interferon expression, is also a promising target for cancer immunotherapies. In the tumor cell, the status of genomic instability, presented by various biomarker panels such as MSI, TMB, and cGAS-STING activation, is vital for evaluating treatment responses to anticancer chemo- and immuno-therapies.
Hepatocellular carcinoma remains among the top death-causing cancers. The main etiologies of HCC include infections of oncogenic viruses and alcohol- and nonalcoholic metabolic liver diseases. This article summarized the updated findings for genomic instability caused by various HCC-associated liver diseases. Viral hepatitis causes liver inflammation and oxidative stress. The viral oncogenic factors such as HBV surface and X proteins specifically interact with several host proteins, inhibiting DNA repair and cell cycle checkpoint. Moreover, the viral integration results in insertional mutagenesis of the host genomes, especially the insertion hotspots, such as TERT, CCNA2, and p53, that are involved in the carcinogenic processes. Furthermore, chronic exposure to genotoxic agents, including alcohol and nonalcoholic metabolic stress, causes mutation accumulation and triggers cell transformation and tumorigenesis. These causes dampen the liver cell in genomic instability-induced carcinogenic inferno.
Though genomic instability renders the cell at high risk to cancer, recent research findings showed that genomic instability serves as a positive biomarker for cancer immunotherapies such as ICI inhibitors and antibodies. Enhanced neoantigen production due to high global mutations stimulates CD8+ T cell cytotoxic activities against the tumor cell. The FDA has approved the genomic instability markers, including MSI and TMB, as predictive biomarkers for positive responses to immunotherapies in treating several cancers. Thus, clinical use of the relevant biomarkers in HCC therapies has yet to be widely accepted. Comprehensive cohort studies must be undertaken to clarify the association of relevant biomarkers with HCC therapeutic efficacies, providing valuable guidelines in clinical practice.
FundingThe project was supported by the Taiwan Ministry of Science and Technology [grant numbers 111-2320-B-006-017, 111-2622-B-006-005, and 112-2811-B-006 -008, to WH; 108-2320-B-041-002, to JHH] and China Medical University, Taichung, Taiwan [grant number CMU111-MF-42, to CFT].
Author contributionsJHH, CFT, and CHC conceptualized and drafted the manuscript. YLC and PJC designed and performed the experiments described in the manuscript. CLH supervised the experiments and manuscript writing. WH coordinated, revised, and submitted the manuscript. All authors read and approved the final manuscript.



