





The in vitro bioactivity of slightly hypereutectic glass-ceramics of the pseudo-binary system diopside (CMS2, CaMgSi2O6)–tricalcium phosphate [Ca3P, Ca3(PO4)2], was tested in a Simulated Body Fluid (SBF) from 7 to 21 days at pH=7.3 and 36.5°C. The materials were synthesized by the petrurgic method, using cooling rates of 0.5, 1 and 2°C/h through the mushy zone. Their microstructure consisted of β-C3Pss (solid solution of CMS2 in Ca3P) primary dendrites in a matrix of CMS2–β-C3Pss lamellar eutectic phase. The dissolution of the interdendritic matrix into the SBF lead to the formation of an interconnected porous structure at the surface of the samples. Subsequently, the β-C3Pss primary dendrites were pseudomorphically converted into (Ca10−yMgy)(PO4)6−z(SiO4)z(OH)2−z (Mg and Si co-substituted hydroxyapatite, HAp). The cavities left by the dissolved matrix were then filled with HAp, and a layer of this phase was formed on the surface of the material. The rate of dissolution of the material's matrix into the SBF, the rate of precipitation of HAp from the solution, the composition of the precipitated HAp, and pH and ionic composition of the SBF during the tests, were all affected by the cooling rate used to synthesize the materials.
La bioactividad in vitro de vitrocerámicas ligeramente hipereutécticas del sistema seudobinario diópsido (CMS2, CaMgSi2O6)–fosfato tricálcico [Ca3P, Ca3(PO4)2], se probó en un fluido fisiológico simulado (FFS) por 7 a 21 días a pH=7.3 y 36.5°C. Los materiales sintetizados por el método petrúrgico, usando velocidades de enfriamiento de 0.5, 1 y 2°C/h a través de la zona de transición sólido/líquido, presentaron una microestructura de dendritas primarias de β-C3Pss (solución sólida de CMS2 en Ca3P) en una matriz de fase eutéctica laminar de CMS2–β-C3Pss. La disolución de la matriz en el FFS creó una estructura porosa interconectada en la superficie de las muestras. Posteriormente, las dendritas primarias de β-C3Pss se convirtieron seudomórficamente en (Ca10−yMgy)(PO4)6−z(SiO4)z(OH)2−z (hidroxiapatita, HAp, co-sustituida con Mg y Si). Las cavidades creadas al disolverse la matriz se llenaron con HAp, y se formó una capa de esta misma fase sobre la superficie del material. Las velocidades de disolución de la matriz en el FFS y de precipitación de la HAp de la solución, la composición de la HAp precipitada, y el pH y la composición iónica del FFS durante las pruebas, fueron afectados por la velocidad de enfriamiento utilizada para sintetizar los materiales.
In order to improve the osseointegration properties (ability to promote the growth of new bone) of biomedical implants, without using mechanically weak porous materials, some dense ceramic materials such as Bioeutectic®, which are capable of generating in situ a porous structure similar to that of bone, have been designed [1]. This material corresponds to the eutectic composition of the binary system wollastonite (CS, CaSiO3)–tricalcium phosphate [C3P, Ca3(PO4)2]. The CS phase contained in it dissolves in contact with a Simulated Body Fluid (SBF), generating in situ an interconnected porous structure of C3P. Then, the C3P porous skeleton transforms pseudomorphically into hydroxyapatite [HAp, Ca10(PO4)6(OH)2]. The subsequent formation of a layer of HAp on the surface of the material creates an adhesion interface between it and the bone tissue [1].
Carrodeguas et al. [2] and García-Páez et al. [3] claimed that the eutectic composition of the system diopside (CMS2, CaMgSi2O6)–C3P, which is considered to be pseudo-binary due to the segregation of SiO2 caused by the formation of the β-C3Pss solid solution [4], is able to exhibit a behavior similar to that of Bioeutectic® in contact with physiological fluids. However, no evidence of the occurrence of a pseudomorphic transformation of β-C3Pss into HAp was presented. It was found that in this material the β-C3Pss phase dissolves preferentially into the SBF, leaving behind a porous surface rich in CMS2. The thickness of the porous layer increases rapidly at the beginning of the soaking time in the SBF, reaching a thickness of ∼20μm during the first 7 days. Subsequently, the increase in the thickness of the porous layer becomes much slower, reaching a value of ∼23μm after 20 days of soaking. Later, the CMS2 partially dissolves into the SBF and the porous surface of the material is coated with a layer of a HAp-like phase. It is hypothesized that the microstructure generated in the material during its soaking in the SBF is controlled by the solubility of the β-C3Pss phase, which in turn is promoted by the presence of CMS2. The dissolution of the β-C3Pss phase modifies the chemistry and topography of the material surface, leading to an in situ formation of an interconnected porous structure of CMS2, which may be expected to promote bone growth.
In the present study, we report the in vitro bioactivity of a glass-ceramic biomaterial synthesized using the so-called “petrurgic method” [5], which has a nominal composition of 61% CMS2–39% C3P (throughout this work, compositions are given in weight percent, unless otherwise specified). This material is of a slightly hypereutectic composition in the pseudo-binary system CMS2–C3P [4], and its microstructure consists of long β-C3Pss skeletal dendrites in a matrix of CMS2–β-C3Pss lamellar eutectic phase, which presumably contains also a small amount of segregated CaO–SiO2 glass. This material is also capable of behaving similarly to Bioeutectic® in contact with a SBF. However, its microstructure and in vitro bioactivity behavior are significantly different from those shown by the eutectic composition of the same pseudo-binary system studied by Carrodeguas et al. [2] and García-Páez et al. [3], which was synthesized by a solid-state reaction process in both cases.
Experimental procedureThe detailed procedure used for the synthesis of the studied glass-ceramic biomaterials is reported in a companion paper [5]. Briefly, a stoichiometric mixture of CaCO3, (NH4)2HPO4, SiO2, and (MgCO3)4·Mg(OH)2·5H2O reagent-grade chemical precursors was prepared and homogenized, and then it was melted at 1450°C/2h in a platinum crucible, using a heating rate of 5°C/min. Then, the melt was cooled down to 1320°C (slightly above the liquidus temperature of the material) at a rate of 3°C/min. Subsequently, the melt was cooled through the mushy zone, within which the newly formed primary solid phase coexists at equilibrium with the remaining liquid during solidification of the material, at a rate of 0.5, 1 or 2°C/h. Upon reaching the eutectic point of the system (1300°C), at which solidification ends, the cooling rate was changed back to 5°C/min and the solidified material was cooled down to room temperature within the crucible inside the furnace. Subsequently, the synthesized glass-ceramic materials were subjected to in vitro bioactivity tests. The specimens used for these tests were taken from the central zone of the crucible, away from its walls. The in vitro bioactivity tests were performed by soaking small pieces of the glass-ceramic materials weighing ∼0.3g for 7, 14 or 21 days in 100ml of corrected SBF (usually denoted in the literature as “c-SBF”, but hereafter referred to simply as SBF) reported by T. Kokubo and H. Takadama [6], which mimics the ionic composition of human blood plasma. The samples were soaked in the SBF inside polyethylene bottles, at a pH of 7.3. Then, the sealed bottles were placed inside an incubator oven (Fisher Scientific Isotemp 650D) at 36.5°C. The bioactivity tests were performed without stirring, and without periodic replenishment of the SBF.
At the end of the period of soaking in the SBF, the substrates were rinsed with deionized water and left to dry inside a glass desiccator, to be characterized later by X-Ray Diffraction (XRD) and Scanning Electron Microscopy (SEM) The XRD analyzes were carried out directly on the surface of the substrates, without any further preparation, using monochromatic CuKα radiation in a Philips X’Pert 3040 apparatus, with a scanning speed of 0.03°/s in the 2θ range from 20° to 50°. The surface and the cross-section of the materials were analyzed on the SEM. The surface of the samples was analyzed after coating it with a graphite film using a JEOL JEE-400 vacuum evaporator, without any further preparation. In contrast, to observe the cross-section of the samples, these were mounted on slow setting epoxy resin, cutting them later with a diamond disk, and proceeding to their ceramographic preparation using standard techniques, with a final mirror polish using diamond paste with successive particle sizes of 3, 1 and 0.5μm. Finally, the latter samples were cleaned, dried and coated with a graphite film. For the observation on the SEM, a Philips XL30 ESEM device with an acceleration voltage of 20kV and a working distance of 10mm was used.
After taking the specimens out of the SBF at the end of the in vitro bioactivity tests, the pH of the solutions was measured using a Thermo Scientific™ Orion™ apparatus, with an accuracy of ±0.002pH, and which was calibrated before each measurement with buffer solutions with pH=4, 7 and 10. Then, the used SBF solutions were stored under refrigeration, for further chemical analysis. This analysis was carried out by Inductively Coupled Plasma atomic emission spectroscopy (ICP) in order to determine the change in the ionic concentration of Ca, P, Si and Mg in the SBF, as a function of soaking time for the different substrates used.
Results and discussionIn vitro bioactivity of the glass-ceramic materialsAccording to Fig. 1, it was observed that from the beginning of the in vitro bioactivity tests a gradual dissolution of the matrix of the material into the SBF took place, while the dendrites of the primary phase remained undissolved as bars protruding from the attacked surface of the material, between which an interconnected porous structure was formed. The continuous dissolution of the matrix lasted for as long as there were no significant obstructions preventing the penetration of the solution into the formed cavities. For the case of the cooling rate of 0.5°C/h, the dissolution depth was ∼104μm and ∼184μm, while for the cooling rate of 1°C/h it was ∼85μm and ∼180μm, and for the cooling rate of 2°C/h it was ∼83μm and ∼191μm, after 7 and 21 days of soaking in the SBF, respectively. Therefore, the initial dissolution rate of the matrix was much faster for the lowest cooling rate than for the other two cooling rates used. However, after 21 days of soaking in the SBF the dissolution depth of the matrix was comparable for the three cooling rates used.

The fact that the matrix started to dissolve into the SBF after relatively short soaking times, while the dendrites of the primary phase did not, can be attributed mainly to the large size of the crystals of the latter phase, as well as to the partial substitution of Mg for Ca in C3P, which considerably diminishes the solubility of this phase in water [7], as well as in aqueous saline solutions [8]. However, it is known [9] that the partial substitution of Si for P decreases the stability of the β-C3P phase. Therefore, our results suggest a predominance of the effect of the first ionic substitution over the second one on the solubility of the β-C3Pss primary phase into the SBF.
While the primary dendrites of the β-C3Pss phase, which were quite large, did not dissolve in the SBF, the lamellae of this same phase present in the eutectic phase in the matrix of the material did dissolve in said solution, which was probably due to its larger surface area associated with its finer size, when compared with the primary dendrites [10]. A synergistic effect could also occur between the solubility of CMS2, β-C3Pss and CaO-SiO2 phases coexisting in the matrix of the material, since it is known that the solubility of the first two phases increases when both of them dissolve simultaneously into the SBF, according to a thermodynamic simulation carried out by García-Páez et al. [3]. Besides, it is also well-known that the glassy phases in general tend to be very reactive and soluble in the SBF.
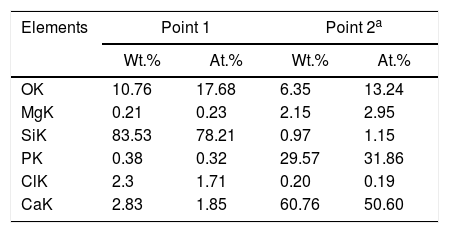
However, with the course of time of soaking in the SBF, the gaps left between the dendrites by the dissolution of the matrix began to be filled gradually, first with what appeared to be a silica gel and later with HAp that precipitated from the solution. The silica gel was detected as a SiO2-rich phase in the samples extracted from the SBF after short soaking times. The results of the EDS analyses carried out for this phase formed in the dissolution zone, as well as for a protruding dendrite, are given in Table 1 for a sample cooled at 0.5°C/h through the mushy zone, after soaking it in SBF for 7 days. The analyzed points are indicated in Fig. 1. Point 1 corresponds to the silica gel and point 2 corresponds to the dendrite. As can be seen, the silica gel is basically composed of SiO2, while the dendrite corresponds to Mg and Si co-substituted HAp, as discussed in detail later.
Compositions (%) determined by EDS analyzes on the SEM of a sample cooled at 0.5°C/h through the mushy zone, after soaking it in SBF for 7 days (analyzed points are indicated in Fig. 1; point 1 corresponds to silica gel formed in the dissolution zone and point 2 to a protruding dendrite).
| Elements | Point 1 | Point 2a | ||
|---|---|---|---|---|
| Wt.% | At.% | Wt.% | At.% | |
| OK | 10.76 | 17.68 | 6.35 | 13.24 |
| MgK | 0.21 | 0.23 | 2.15 | 2.95 |
| SiK | 83.53 | 78.21 | 0.97 | 1.15 |
| PK | 0.38 | 0.32 | 29.57 | 31.86 |
| ClK | 2.3 | 1.71 | 0.20 | 0.19 |
| CaK | 2.83 | 1.85 | 60.76 | 50.60 |
On the other hand, the HAp formed in that region was clearly observed in the case of the samples soaked for 21 days in the SBF, especially in the one crystallized at 0.5°C/h. The silica gel contains silanol groups (Si–OH) on its surface [11]. These groups can act as heterogeneous nucleation sites for HAp in the interdendritic zone, where the material's matrix has dissolved into the SBF. This is further promoted by the supersaturation in Ca and P of the solution located at that zone [10]. However, it is known [12] that the true determinant factor for the precipitation of HAp is the abrupt elevation of the pH of the SBF, up to a range of 9–10.5, which takes place at the dissolution front due to the ion exchange occurring between the H3O+ of the SBF and the Ca2+ and Mg2+ ions contained in the material's matrix. At this pH range, part of the hydrogel dissolves into the SBF in the form of SiO32−, with the subsequent precipitation of HAp. By following this process, eventually all previously formed silica gel should be consumed for the formation of HAp.
Simultaneously, the occurrence of the pseudomorphic transformation of the β-C3Pss dendrites into a Mg and Si co-substituted HAp was observed. This was deduced based on the observation that the dendrites found in the material crystallized at 0.5°C/h had a (Ca+Mg)/(P+Si) molar ratio equal to 1.5 prior to soaking it in the SBF, as reported in the companion paper [5]. However, the value of this molar ratio increased to 1.62 in the dendrites protruding from the sample surface after soaking for 7 days in the SBF, see Table 1. Therefore, it was concluded that while in the first case the dendrites are composed of β-C3Pss, in the second case they are composed of a Mg and Si co-substituted HAp.
The occurrence of this phenomenon is also supported by the results previously reported by other researchers. For example, de Aza et al. [1] reported the occurrence of a pseudomorphic transformation of C3P into HAp for the case of Bioeutectic® in contact with a SBF. Likewise, LeGeros et al. [10], in in vivo bioactivity tests carried out in rabbits, reported a similarity in the Ca/P ratio of the new bone with respect to that of the Mg-substituted β-C3P implants, which suggests the occurrence of the transformation of the latter material into HAp similar to the mineral phase of bone, which took place by a partial dissolution and reprecipitation process, similar to the one previously reported for other calcium phosphates.
Putnis [13] presented a series of important considerations on the factors that provoke the occurrence of pseudomorphic transformations in mineral systems. This author mentions that in order to preserve the external morphology of the original phase, the dissolution and precipitation reactions must be tightly coupled at the interface formed between the original and the product phases, with a well-defined reaction front, in addition to being well-coupled in time. In order for the fluid to maintain contact with the reaction front, the product phase must develop intracrystalline porosity and permeability. The controlling stage of the process kinetics is the rate of dissolution, and there is a low activation energy for nucleation. The dissolution of the original phase results in the formation of a supersaturated solution with respect to the product, in the so-called “boundary layer” formed at the interface with the fluid. Depending on the transport speed of the species in the solution, to and from the interface, even the dissolution of a monolayer of the original phase may oversaturate the boundary layer with respect to the product phase. The nucleation of the product phase takes place on the surface of the dissolving original phase, which makes that the external surfaces of the new phase and the original crystal correspond to each other. If the solubilities of the original and product phases are very different, even a small amount of dissolution will result in a highly supersaturated fluid, with which nucleation will be rapid, even if there is no crystallographic relationship between the two solid phases.
The previous considerations of Putnis [13] suggest that in the present work the lack of mechanical stirring of the SBF during the in vitro bioactivity tests could play an important role on the occurrence of the pseudomorphic transformation of the β-C3Pss dendrites into HAp.
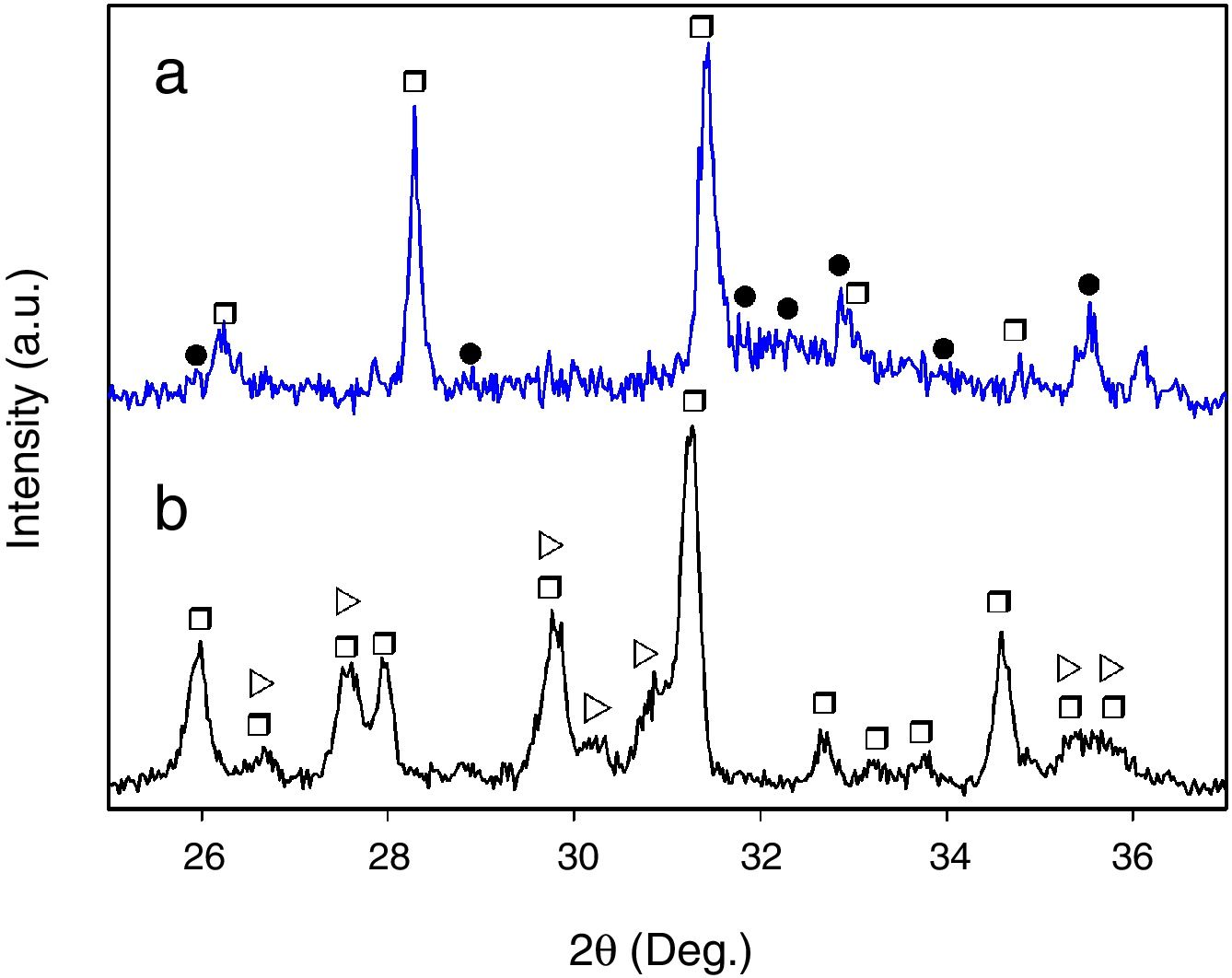
Finally, when Ca and Mg that were dissolved from the CMS2 and β-C3Pss phases diffused toward the SBF located far away from the dissolution interface, and when the solution was supersaturated in Ca and P, precipitation of Mg and Si co-substituted HAp occurred on the surface of the material, directly from the solution. This can be clearly seen in the Ca and P elemental distribution maps shown in Fig. 1 for the material crystallized at 0.5°C/h, as well as in Fig. 2 for all cooling rates used. The EDS spectra shown in the latter figure indicate that the HAp precipitated on the surface of the samples at this stage is Ca-deficient, and that it is also partially substituted with Mg and Si. The micrographs taken at higher magnification shown in the same figure indicate that the precipitated HAp has the typical rosette-shaped globule morphology observed in other studies [14–16]. After 21 days of soaking in the SBF, all the substrates presented a homogeneous HAp coating on their surface. The cracks observed in some cases in the HAp layer are due to volume contraction that took place during natural drying of the samples after the in vitro bioactivity tests [17]. Fig. 3 shows the XRD patterns corresponding to a sample crystallized at 1°C/h, without soaking in the SBF (Fig. 3b), and after 21 days of soaking in the SBF (Fig. 3a). In the latter XRD pattern, it can be seen that a series of low intensity peaks appear in the range of ∼31.5° to ∼34°, which do not appear in the sample without soaking in the SBF, and which, according to the published literature [18,19], correspond to Mg and Si partially substituted HAp.
![Formation of a HAp layer on samples crystallized using different cooling rates through the mushy zone, as a function of time of soaking into the SBF. The EDS spectra of the HAp globules are also shown [a) 2, b) 1, and c) 0.5°C/h].](https://static.elsevier.es/multimedia/03663175/0000006000000002/v1_202104020823/S0366317520300170/v1_202104020823/en/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNd3TCEZuNxhudow/+3Hrh9WJc+mtnYSSPL9K6mfMWVj1pPzg67xfR0+MXeGtM68Mq7kAJu9aQ+n1TzC3T7zV1SOpPqjxx+PmoRLM0Sl+pciQq1WBrzcTWZechVKv/Zg6BsiOpvtu0FOzplXqSugA41Wvu/AlaqhPMjTHKhr6+lWEW43LKE+VTfcxqTVirLxXZDFGi6dKGBer5svzgxtXyI1j8upF2i7toedQt5ZUmqleviseXFAD/wx1y37Cf4z/t4= "Formation of a HAp layer on samples crystallized using different cooling rates through the mushy zone, as a function of time of soaking into the SBF. The EDS spectra of the HAp globules are also shown [a) 2, b) 1, and c) 0.5°C/h].")
 After 21 days of soaking in the SBF, and b) Without soaking in the SBF. Key: ▶ CMS2 (JCPDS card no. 98-003-0522), □ β-C3Pss (JCPDS card no. 01-070-0682), and ● HAp (JCPDS card no. 01-073-8419).")
The precipitation of HAp inside the voids created by the dissolution of the matrix, as well as on the surface of the samples, eventually closed the access of the solution toward the matrix dissolution front. In some cases, this provoked that a small amount of silica gel and residual porosity remained trapped inside the closed voids, especially by the end of the in vitro bioactivity tests. This can be clearly seen in the Si distribution maps shown in Fig. 1, corresponding to a sample crystallized at 0.5, although this was more accentuated for the case of the fastest cooling rate used (2°C/h). The occurrence of this phenomenon also led to the presence of a remnant unaffected core inside the glass-ceramic pieces, toward the end of the in vitro bioactivity tests.
It should be noted that, as in the case of the CaO-SiO2 glass segregated during the formation of the β-C3Pss phase, the small amount of silica gel that was trapped inside the interdendritic spaces due to the precipitation of HAp does not negatively affect the bioactivity of the materials, since it is well-known [20] that SiO2 possesses good bioactivity properties.
Finally, the β-C3Pss dendrites that were pseudomorphically converted into HAp in contact with the SBF could act as micro anchors, and this could confer good osseointegration properties to the synthesized materials, although this is an aspect that requires further investigation.
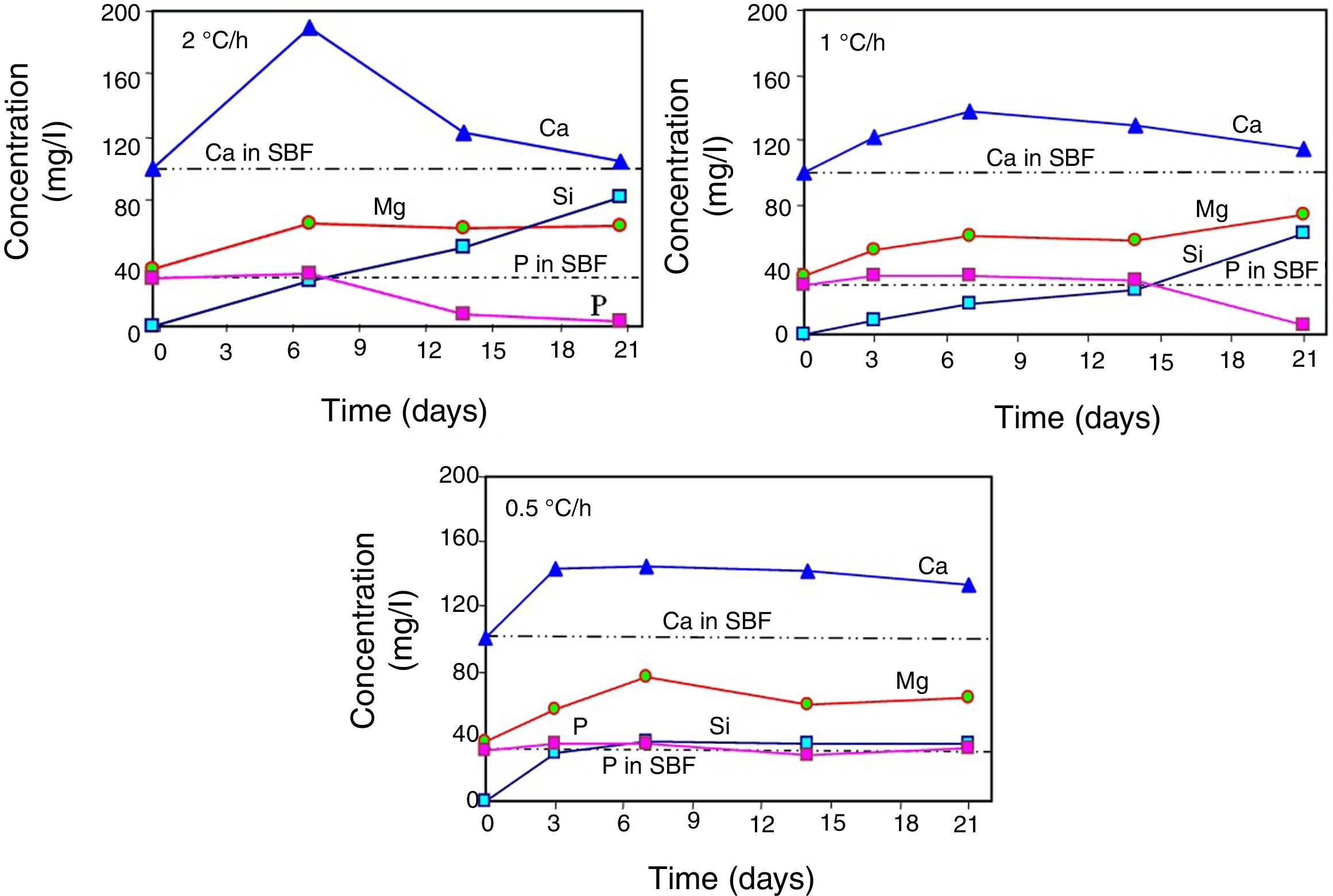
Changes in the ionic composition and pH of the SBF during the in vitro bioactivity testsAccording to Fig. 4, in all cases the concentration of the Ca2+ ions increased in the SBF during the first days of the test, reaching a maximum value after 3 days of soaking for the sample crystallized at 0.5°C/h, and after 7 days of testing for the other two cooling rates used. The highest maximum concentration was achieved for the material crystallized at 2°C/h. From that point onwards and until the end of the test, the concentration of the Ca2+ ions decreased in the SBF, in a faster way with increasing cooling rate. In this manner, for the case of the material crystallized at 2°C/h, after 21 days of soaking the Ca2+ ions reached a concentration level very similar to that of the initial SBF.

Also in all cases, the concentration of Mg2+ ions increased in the SBF during the first 7 days of the test, reaching at this point similar maximum concentrations for the three cooling rates used. From this point onwards and until the end of the test, the concentration of these ions suffered only slight variations.
In the case of the cooling rate of 0.5°C/h, the concentration of Si4+ ions increased rapidly in the SBF during the first 3 days of the test, reaching at that point a level very similar to that of the P5+ ions in the initial solution. From that point onwards and until the end of the test, the concentration of these ions remained practically constant. In contrast, for the other two cooling rates used, the concentration of Si4+ ions increased steadily throughout the test, in a manner almost linear, which was faster with increasing cooling rate. In this way, the highest final concentration of Si4+ ions was achieved in the SBF after 21 days of soaking of the material crystallized at 2°C/h.
In the case of the cooling rate of 0.5°C/h, the concentration of the P5+ ions remained practically constant, at values very close to that of the initial solution, during the whole duration of the test. The same happened until day 14 for the cooling rate of 1°C/h, and until day 7 for the cooling rate of 2°C/h. In these last two cases, at the mentioned point a rapid decrease in the concentration of the P5+ ions started in the SBF, which was more pronounced for the fastest cooling rate used, reaching in both cases a nearly null concentration after 21 days of testing.
The decrease observed in the concentration of Ca2+ ions in the SBF after 7 days of soaking, for all cooling rates used, as well as in the concentration of P5+ ions after a certain soaking time, which decreases with increasing cooling rate, are both associated with the onset of precipitation of HAp from the solution. This moment corresponds to the point at which supersaturation is reached in the SBF for these ions. At the beginning of the tests, the amount of Ca2+ ions released from the material's matrix into the SBF was faster with increasing cooling rate. Thus, supersaturation of the solution with these ions was reached sooner for the case of the fastest cooling rate used. Therefore, it was in this case that the HAp layer began to form sooner, and faster, on the surface of the glass-ceramic materials. This occurred despite the fact that the matrix dissolved into the SBF at a higher rate in the case of the materials crystallized at 0.5°C/h. This is because, at relatively short soaking times, the latter materials released a smaller amount of Ca2+, and a larger amount of Si4+, into the SBF, when compared with the materials crystallized at higher cooling rates. This in turn was due, as already mentioned, to the highest proportion of CaO-SiO2 glass present in the matrix of the material crystallized at the slowest cooling rate used. It should be mentioned that the HAp precipitation did not modify the pH of the solution bulk appreciably. In fact, such precipitation was further favored by the fact that the solution was alkaline at the time it took place, and HAp is the most stable calcium phosphate under such conditions [21].
For all heating rates studied, the pH of the solution bulk slightly increased from 7.3 to ∼7.6 between the first day and the 21st day of immersion in the SBF. This was attributed to the ion exchange occurring between the SBF and the substrate, as well as to the dissolution of the material's matrix into the solution [1]. This in turn led to a gradual increase in the concentration of Ca, Mg and Si in the solution, as already mentioned. For the case of the cooling rate of 0.5°C/h, there was a greater initial dissolution of the matrix, which probably was due to a likely higher proportion of the segregated CaO-SiO2 glass present in it [5]. It is worth mentioning that it is known [12] that precipitation of HAp requires an increase in pH to 9–10.5 at the dissolution front. Thus, although the pH of the solution bulk was only slightly alkaline, the fact that HAp precipitation was observed indicates that sufficiently high pH values were reached at the dissolution front.
The fact that the HAp layer began to form sooner, and faster, on the surface of the glass-ceramic materials that were crystallized with the fastest cooling rate used, explains why for this case the amount of silica gel and residual porosity that remained trapped inside the interdendritic voids was the largest at the end of the bioactivity tests. In other words, in this case the access to the voids was blocked sooner by HAp precipitated on the surface of the samples, with respect to the materials synthesized using the other two cooling rates.
This also implied that, although Ca, Mg, Si and P were constantly released from the matrix of the material into the SBF during the entire duration of the tests, this occurred at a decreasing rate as time passed, due to the gradual blockage of the void access for the incoming liquid, caused by the HAp precipitation. The pseudomorphic transformation of the β-C3Pss dendrites into HAp was another source of Mg and Si for the SBF (reaction (5)). The HAp precipitation, and the pseudomorphic transformation of the β-C3Pss dendrites into HAp, rapidly consumed any amount of Ca and P released from the matrix of the material, with significant amounts of both ions initially present in the SBF also consumed in this process. This caused a gradual depletion of the SBF in Ca and P, which was more accentuated as the cooling rate increased. The fact that in all cases the concentration of Mg2+ increased during the first 7 days of soaking in the SBF, remaining constant from that point onwards until the end of the test, together with the fact that the concentration of Si4+ increased continuously throughout the test, was associated with the precipitation of Mg and Si partially substituted HAp, with the first substitution being the most important one, especially for the case of the samples crystallized with the fastest cooling rate used. The fastest rate of formation of the HAp layer on the surface of the latter samples implied a relatively short time of incorporation of Si4+ into this layer.
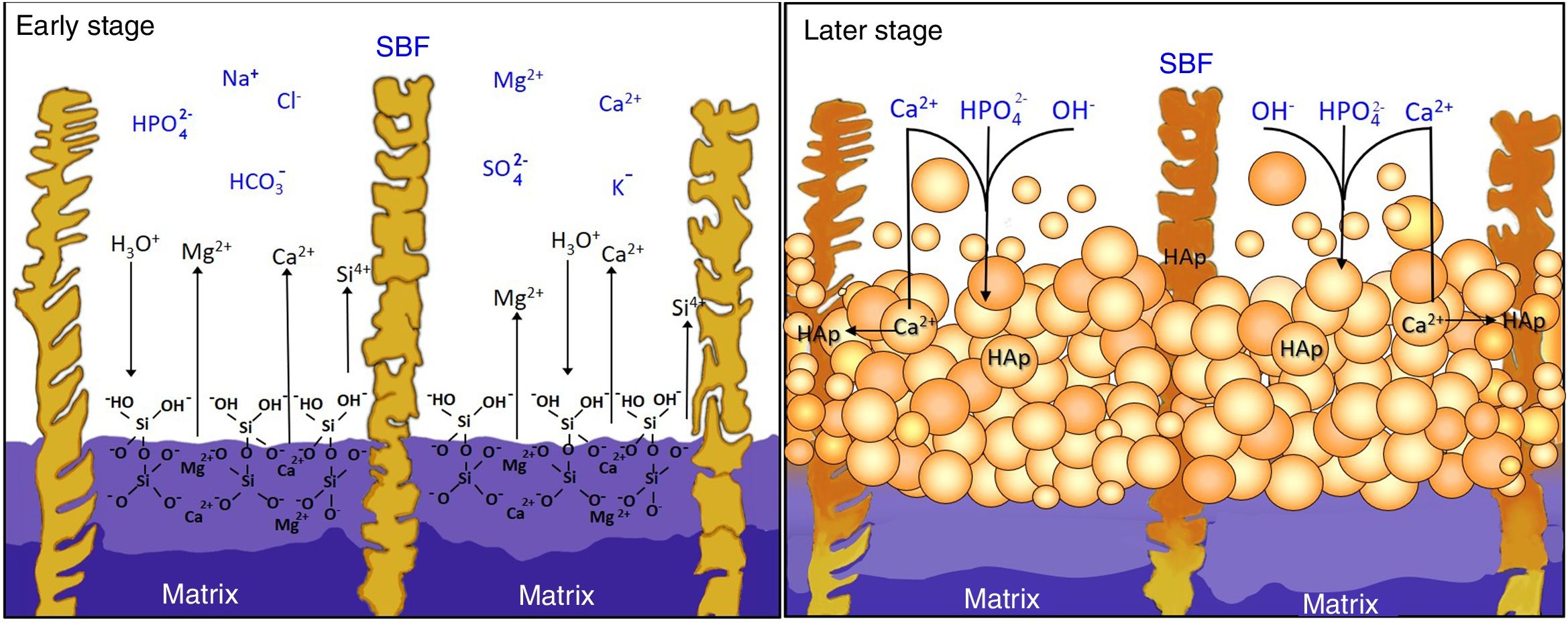
Reaction mechanism during the in vitro bioactivity testsThe hypothesized mechanism of formation of HAp at the surface of the synthesized materials during the in vitro bioactivity tests is summarized in Fig. 5.

In the proposed mechanism, at the beginning of the in vitro bioactivity tests an ion exchange occurred between H3O+ coming from the SBF, and Ca and Mg contained in the CMS2 present in the material's matrix (reaction (1)) [3]. This also caused Si from CMS2 to dissolve into the solution in the form of HSiO3−. Simultaneously, the segregated CaO-SiO2 glass and the β-C3Pss phase present in the matrix were also dissolved into the SBF. The CaO-SiO2 glass did it through reaction (2), while the β-C3Pss phase did it through the action of the H2PO4− ions present in the solution (reaction (3)).
This caused an increase in the concentration of Ca, Mg and Si in the solution, as well as an increase in the pH of the latter, especially at the dissolution front. When a pH level between 8 and 11 was reached at this front [12], an equilibrium was established between the HSiO3− ions and silicic acid [H4SiO4 or Si(OH)4] [22], which resulted in the formation of a certain amount of this acid in the solution. Silicic acid contains four silanol groups, and when it is hydrolyzed it can undergo a condensation reaction (polymerization) to produce a silica gel, according to reaction (4)[23]:
The gaps left by the dissolution of the material's matrix began to be gradually filled with HAp. The silanol groups formed on the surface of the silica gel during condensation [11] could act as heterogeneous nucleation sites for HAp, which was also promoted by supersaturation of the solution in Ca and P [3]. However, it is known [12] that the true determinant factor for HAp precipitation is the increase in pH to 9–10.5 occurring at the dissolution interface due to the ion exchange taking place between H3O+ from the SBF and Ca2+ and Mg2+ from the material. The dissolution of β-C3Pss into the SBF, reaction (3), also contributed to this increase in pH. In the mentioned pH range, part of the hydrogel was dissolved into the SBF in the form of SiO32−, with the subsequent precipitation of HAp.
At the same time, the pseudomorphic transformation of the β-C3Pss dendrites into Mg and Si co-substituted HAp took place through reaction (5):
Lastly, when Ca and Mg dissolved from the CMS2 and β-C3Pss phases diffused toward the SBF located far away from the dissolution interface, and when the solution was supersaturated in Ca and P, precipitation of Mg and Si co-substituted HAp took place on the surface of the material, directly from the solution (reaction (6)).
This eventually closed the access of the SBF into the interdendritic gaps, in such a way that, toward the end of the in vitro bioactivity tests, in some cases a small amount of silica gel and residual porosity remained trapped inside the closed cavities.
One final remark. In order to fully appreciate the figures in color included in this paper, we encourage the reader to see them in the digital version of the work, since they may appear in black and white in its printed version.
ConclusionsFrom the beginning of the in vitro bioactivity tests, the matrix of the material was gradually dissolved into the SBF, while the dendrites of the β-C3Pss primary phase remained undissolved as bars protruding from the attacked surface of the material, between which an interconnected porous structure was formed. Silicon dissolved from the material's matrix into the SBF located at the interdendritic zone led to the formation of a silica gel containing silanol (Si-OH) groups at that site, which acted as heterogeneous nucleation sites for the formation of HAp. Simultaneously, the occurrence of a pseudomorphic transformation of the β-C3Pss dendrites into Mg and Si co-substituted HAp, (Ca10-yMgy)(PO4)6-z(SiO4)z(OH)2-z, was observed. When Ca and Mg dissolved from the CMS2 and β-C3Pss phases contained in the material's matrix diffused toward the SBF located far away from the dissolution front, and when the solution was supersaturated in Ca and P, precipitation of Mg and Si co-substituted HAp occurred on the surface of the material, directly from the solution. The latter phenomenon was accompanied by concomitant changes in the ionic composition and pH of the SBF. It was hypothesized that the β-C3Pss dendrites that were pseudomorphically converted into HAp in contact with the SBF could act as microscopic anchors, which could potentially confer good osseointegration properties to the synthesized biomaterials. Finally, the rate of dissolution of the material's matrix into the SBF, the rate of precipitation of HAp from the solution, the composition of the precipitated HAp, and the ionic composition of the SBF during the in vitro bioactivity tests, were all affected by the cooling rate used through the mushy zone during crystallization of the samples. The material with the best performance in vitro was the one crystallized at 0.5°C/h, since it showed the fastest initial dissolution rate of the matrix in contact with the SBF, and the cavities formed in it by said dissolution were better filled with precipitated HAp after 21 days of soaking in the fluid.
Author contribution statementJ. López-Cuevas ensures that all authors are included in the author list, its order has been agreed by all authors, and that all authors are aware that the paper was submitted. He originated the main idea of the research work and supervised the conducted experimental work, participating in every aspect of it. He also wrote the draft paper and prepared the final manuscript.
C.M. Lopez-Badillo carried out the experimental work, and contributed to the analysis and discussion of the results and writing of the paper.
J. Méndez-Nonell co-directed the experimental work, assisted in every aspect of the experimental and characterization work, and supervised the final manuscript.
Conflict of interest statementThe authors declare that they have no conflict of interest.
J.L.C. thanks SEP and Cinvestav for the funding granted for the realization of this work (Cinvestav's Scientific Research and Technological Development Fund, Project No. 252). C.M.L.B. also thanks CONACYT for the scholarship granted for the completion of her Master of Science studies at Cinvestav-Saltillo, México. This paper is based on part of her M.Sc. thesis work.

