









A mixture of olivine and (NH4)2SO4 was reacted in the solid state at 300–500°C for 1h. This was followed by mechanical activation of the reacted material for 4–16h in a laboratory attrition mill. Si and Fe were removed from the milled material and Mg was extracted from it as brucite [Mg(OH)2]. CO2 absorption tests were carried out for 24–72h at 50–200°C, using the obtained brucite suspended in deionized water. In these tests, the brucite was completely transformed into nesquehonite (MgCO3·3H2O) at 50°C/24h. Then, nesquehonite was completely transformed into hydromagnesite [Mg5(CO3)4(OH)2·4H2O] at 100°C/24h. Additional CO2 absorption tests were carried out using the obtained hydromagnesite, but this time under a flow of CO2 with a relative humidity of 70–80%, at the same temperatures mentioned before, and with an absorption time of 3 or 6h. It was found that at 200°C/6h hydromagnesite was almost completely transformed into an amorphous hydrated magnesite. In terms of the CO2 capture, the best results were obtained at 50°C/24h with brucite suspended in water. With an industrial-scale process, the nesquehonite produced could be used to manufacture various materials useful for the construction industry.
Una mezcla de olivina y (NH4)2SO4 se hizo reaccionar en estado sólido a 300-500°C por 1h. Esto fue seguido de activación mecánica de la mezcla por 4-16h en un molino de atrición. Se eliminó el Si y el Fe del material molido y se extrajo el Mg como brucita [Mg(OH)2]. Se llevaron a cabo pruebas de absorción de CO2 por 24-72h a 50-200°C, utilizando suspensiones de brucita en agua desionizada. En estas pruebas, la brucita se transformó completamente en nesquehonita (MgCO3·3H2O) a 50°C/24h. Luego, esta se transformó completamente en hidromagnesita [Mg5(CO3)4(OH)2·4H2O] a 100°C/24h. Se llevaron a cabo pruebas adicionales de absorción de CO2 utilizando la hidromagnesita obtenida, por 3 o 6h bajo un flujo de CO2 con una humedad relativa del 70-80%, a las mismas temperaturas mencionadas anteriormente. Se encontró que a 200°C/6h la hidromagnesita se transformó casi completamente en una magnesita hidratada amorfa. En cuanto a la captura de CO2, los mejores resultados se obtuvieron a 50°C/24h con brucita suspendida en agua. Con un proceso escalado a nivel industrial, la nesquehonita producida podría ser utilizada para fabricar diversos materiales útiles para la industria de la construcción.
At the present time, the greenhouse effect is without doubt the most important environmental problem faced by humanity. It is well known that one of the main gases that contribute to this effect is carbon dioxide (CO2) [1–4]. According to the World Meteorological Organization (WMO), the increase in the concentration of this gas in the atmosphere has caused global warming in a more accelerated manner than expected [5]. Under these circumstances, the International Energy Agency (IEA) has pointed out that it is essential to develop CO2 capture and storage technologies that allow reducing as much as possible the emissions of this gas into the atmosphere [6,7]. Among the different technologies that have been developed for this purpose, the so-called “mineral carbonation” stands out, which is based on the exothermic chemical reaction between a metal oxide such as MgO or CaO with CO2 to form solid carbonates that are stable for long periods of time [8]. Therefore, this technique provides a permanent CO2 removal method, producing stable carbonates that are benign for the environment [9,10]. In particular, mineral materials with a high content of Mg and Ca are considered as one of the best options to carry out this carbonation process, due to their great affinity for CO2 and since the chemical reaction that occurs between them and the gas is very simple; another of their main advantages is their low cost and wide availability. Many authors have considered the olivine mineral [(Mg,Fe)2SiO4] as a good candidate for the capture of CO2 due to its great abundance in nature (it constitutes about 8% of the earth's crust and most of the oceanic crust), as well as due to its low cost, environmentally friendly characteristics, high dissolution rate and high capture capacity (it is the silicate containing the lowest proportion of Si), it does not contain aluminum (thus, it does not generate the formation of clays that affect its reactivity), and it has a certain basicity, contributing to the neutralization of H2CO3 formed when CO2 reacts with H2O [11,12]. One of its main advantages is that it does not require any pre-treatment at high temperature to absorb CO2. Kwon et al. [13,14] showed that each gram of olivine can absorb up to 0.18 grams of CO2, requiring only at least 200°C in the presence of water vapor, without subjecting the mineral to any kind of pretreatment. The CO2 capture capacity of this mineral can be further improved by subjecting it to a mechanical activation process (prolonged high energy milling), which considerably increases its surface area, making it more reactive [15]. Prolonged milling causes a loss of crystallinity in the material, which then becomes amorphous [16,17]. It can also result in the formation of new phases. This powder processing technique has been applied to various metallic, ceramic, polymer and composite materials, in order to obtain in them characteristics such as high specific surface area, high homogeneity and a large number of surface defects [18].
Olivine can also be used indirectly for CO2 capture. For instance, this mineral can be reacted with (NH4)2SO4 in the solid state to obtain MgSO4. Then, the latter compound is dissolved in water, and the precipitation of brucite [Mg(OH)2] from the solution is provoked by the addition of NH4OH to it. Subsequently, the precipitated brucite can be used for CO2 capture [19]. In order to increase the reactivity of the system, the mechanical activation of olivine-(NH4)2SO4 mixtures, prior to their reaction in the solid state, has been reported [20]. However, until now the process in which the reaction is first carried out in the solid state, and then the reacted mixture is subjected to mechanical activation, has not been studied. This is the option that has been explored in the present work. It was intended to promote the decomposition, through the action of a prolonged high-energy milling, of any unreacted (NH4)2SO4 that may have been left over after this compound reacted with olivine in the solid state. This was based on the relatively low temperature at which (NH4)2SO4 decomposes [21], which makes feasible its decomposition by mechanical milling. The decomposition of unreacted (NH4)2SO4 is desirable considering the fact that this compound can negatively interfere with the precipitation process of brucite.
Experimental procedureInitial oreOlivine ore from the Santo Domingo volcanic field, San Luis Potosí, Mexico, was used. Its chemical composition was: 42.71% MgO, 42.25% SiO2, 8.84% Fe2O3, 2.14% Al2O3, 0.943% CaO, 0.77% P2O5, 0.43% Cr2O3 and 0.425% NiO, with 1.22% of ignition losses (LOI) determined by thermogravimetry at 950°C/1h. The mean particle size of the initial mineral was ∼30μm. Throughout this work, all percentages are given by weight, unless otherwise specified.
Solid state reaction of olivine with (NH4)2SO4The brucite used in the CO2 absorption tests was synthesized based on the procedure reported by Nduagu [19], in which this compound is obtained by precipitation from an aqueous solution of MgSO4. This in turn is synthesized by reacting the olivine mineral with (NH4)2SO4 (in this work this compound was reagent grade from Sigma-Aldrich), according to reaction 1:
To carry out reaction (1), a mixture of approximately 30g of olivine and (NH4)2SO4 was prepared, both in stoichiometric proportions, subsequently adding 50ml of deionized water to it. This led to the dissolution of the ammonium sulfate in the water, while the olivine particles remained in suspension. Then, the suspension was placed in a porcelain crucible with a capacity of 200ml, which was introduced into a Thermolyne F62700 electric furnace, at a temperature of 300, 400 or 500°C for 1h.
Mechanical activation of reacted materialsThe reacted mixtures were mechanically activated in a Teflon-lined closed chamber laboratory attrition mill operated at ∼1600rpm. Milling was carried out in air at room temperature, for times of 0, 4, 8 and 16h, employing 8mol.% MgO-partially-stabilized ZrO2 balls with a diameter of 1mm as milling media. A ball/load mass ratio of 10/1 was used, employing 10g of sample and 150ml of ethanol as dispersant, occupying in total approximately two-thirds of the volume of the mill container.
Removal of Si and Fe from the mechanically activated materialsIt is known [22] that the formation of MgSO4 favors the precipitation of brucite, while Si and Fe have a retarding effect on it. Therefore, it was sought to remove as much as possible of the latter two elements from the mineral. For this purpose, a solution rich in Mg, Si and Fe was obtained. This was done by mixing the mechanically activated powder with 100ml of deionized water by magnetic stirring at 80°C for 30min; then the solution was filtered. The filtered solution had a pH=1. The remaining solid was stirred again with water and the process was repeated. Subsequently, NH4OH (reagent grade compound from Sigma Aldrich) was added dropwise to the previous solution, until a pH=8 was reached, in order to cause the precipitation of Fe and Si dissolved in it, in the form of iron oxide and silica, thus facilitating the extraction of both phases by subsequent conventional filtration.
Precipitation of brucite from the solution free from Si and FeThe production of brucite was carried out from the solution free from Si and Fe, to which a small volume of NH4OH was added to increase its pH from 8 to 11. This allowed magnesium ions to interact with the added ammonium hydroxide, precipitating brucite [23]. This happened according to reaction 2:
Then, the precipitate was washed with deionized water and dried at 100°C for 6h.
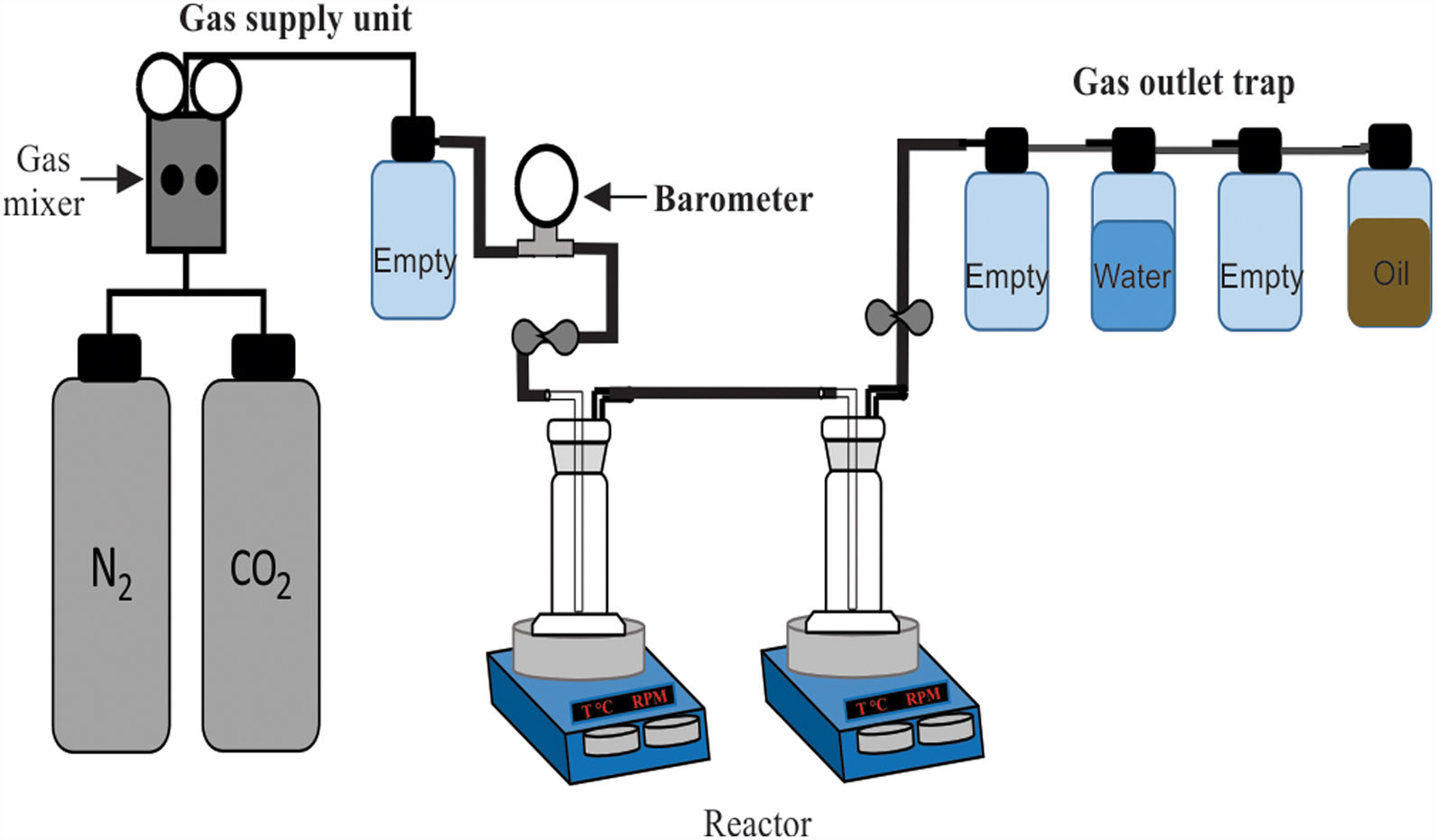
CO2 absorption testsTo carry out the CO2 absorption tests for the synthesized brucite, an experimental equipment consisting of three main parts was used, which are as follows (Fig. 1): (1) a gas supply unit, (2) a reactor, and (3) an oil trap for the gas outlet. The gas supply unit consists of two N2 and CO2 cylinders, the first one of ultra-high purity and the second one with a purity of 95%, plus a WITT BM-2M gas mixer, capable of handling N2/CO2 mixtures from 100% N2 to 100% CO2, with flow rates of 0–25l/min. The reactor consists of two sealed pyrex glass containers (gas bubbler and washing bottles), which are connected in series and placed on two IKA magnetic hot plate stirrers. To carry out the tests, a 5g-brucite sample was suspended in deionized water inside each of the gas bubblers, and then a flow of 100% CO2 gas was passed through them at a rate of 10ml/min, using a stirring speed of 300rpm for both gas bubblers. The tests were carried out in this way based on the assumption, according to Fricker and Park [24], that in a humid environment the rapid formation of intermediate species of hydrated magnesium carbonates is promoted, which can be transformed into anhydrous magnesium carbonates by increasing humidity, as well as the temperature and/or time of reaction. The oil trap had the function of preventing the entry of air from the environment, in the event of an accidental drop in pressure inside the reactor. The temperature used during the tests was 50, 100, 150 or 200°C, using a heating rate of 10°C/min, with a CO2 absorption time of 24 or 72h.

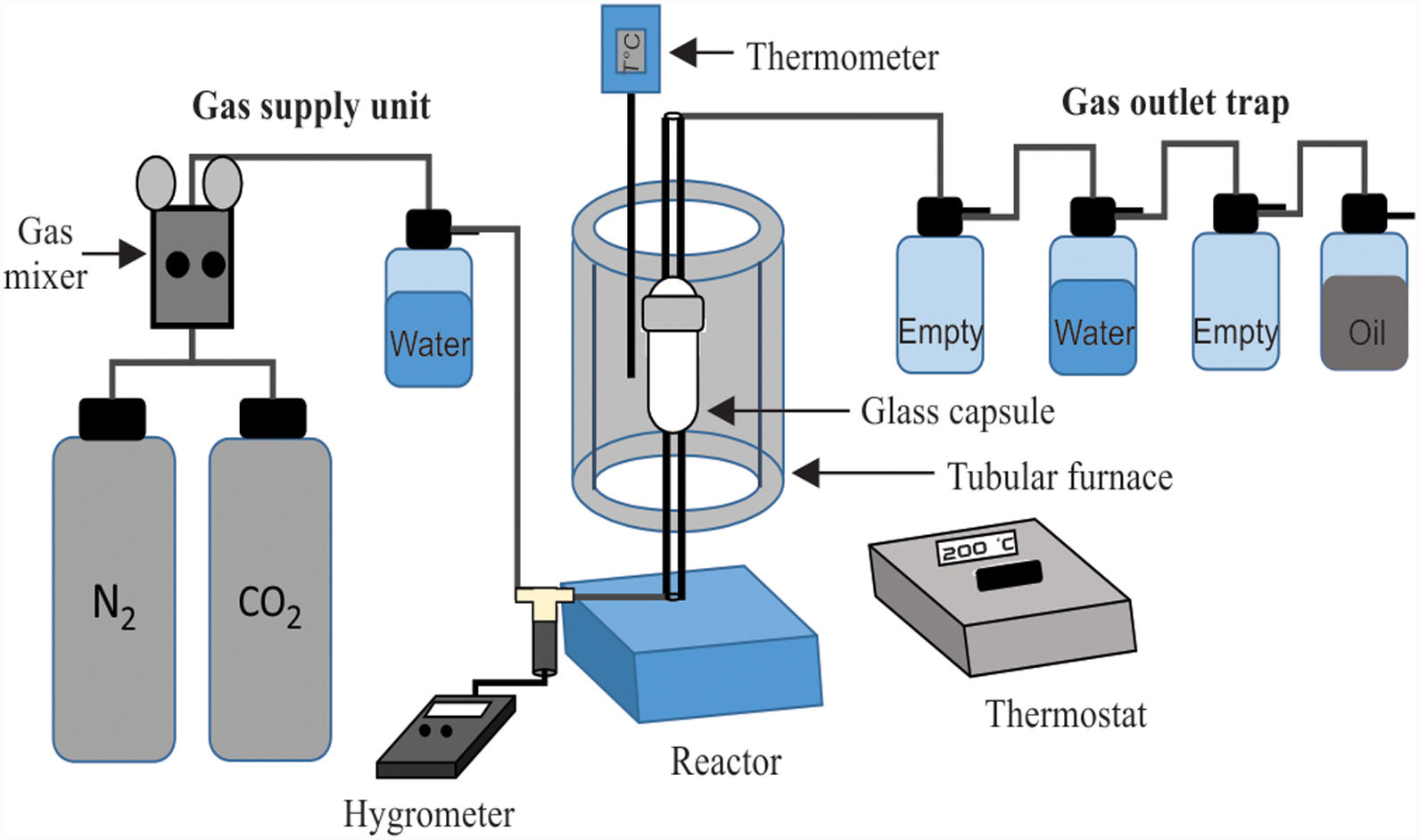
The materials produced in the previous tests were subjected to additional CO2 absorption experiments, for which the experimental equipment shown in Fig. 2 was used. This equipment is similar to that shown in Fig. 1, except for the fact that in this case a different reactor was used, and a gas humidifier (of the type used in hospitals for the humidification of medical oxygen), as well as a Control Company EA200 hygrometer, were placed just before the reactor entrance. This consisted of an oval-shaped pyrex glass capsule, specially manufactured for us, which has a length of 10cm, a diameter of 2cm, and a volume of 32cm3, and whose ends are attached to two glass tubes with 20cm in length and 0.5cm in diameter. The capsule can be separated into two parts for the placement of a powder sample inside it. To carry out the tests, the capsule was opened, and the powder was placed inside its lower part, supported on a stainless-steel wire mesh with an opening of 37μm, which in turn rested horizontally on glass wool. The capsule was then sealed and placed inside a Thermolyne 21,100 vertical tube furnace. The capsule was placed in the center of the furnace tube to ensure a uniform temperature during the tests. A 100% CO2 gas flow was then passed through it at a rate of 10ml/min, with a relative humidity of 70–80%. The temperature used during the tests was 50, 100, 150 or 200°C, using a heating rate of 20°C/min, with a CO2 absorption time of 3 or 6h.
Characterization of powdered materials and aqueous solutions
All powdered materials were analyzed by using the following techniques: (a) X-ray Diffraction (XRD), using Cukα radiation in a Philips X-PERT device, (b) X-ray Fluorescence Spectrometry (XRF), using a BRUKER S4 PIONNER apparatus, (c) Fourier Transform Infrared Spectroscopy (FT-IR), using a Nicolet Avatar 320 device, and (d) Differential Thermal Analysis (DTA) and Thermogravimetry (TGA) using either a simultaneous DTA/TG/DSC TA Instruments SDT Q600 analyzer or a Perkin-Elmer TAC/DX apparatus, in both cases with platinum crucibles and using alumina as reference material, at a heating rate of 10°C/min up to 800°C, and with a continuous flow of argon gas. The particle size distribution was determined by laser dispersion using a Horiba Partica LA-950V2 equipment, employing ethanol for powder dispersion, with the application of ultrasonic stirring for 5min. The liquid solutions were analyzed by Inductively Coupled Plasma Spectroscopy (ICP), using a Perkin Elmer Optima 8300 apparatus.
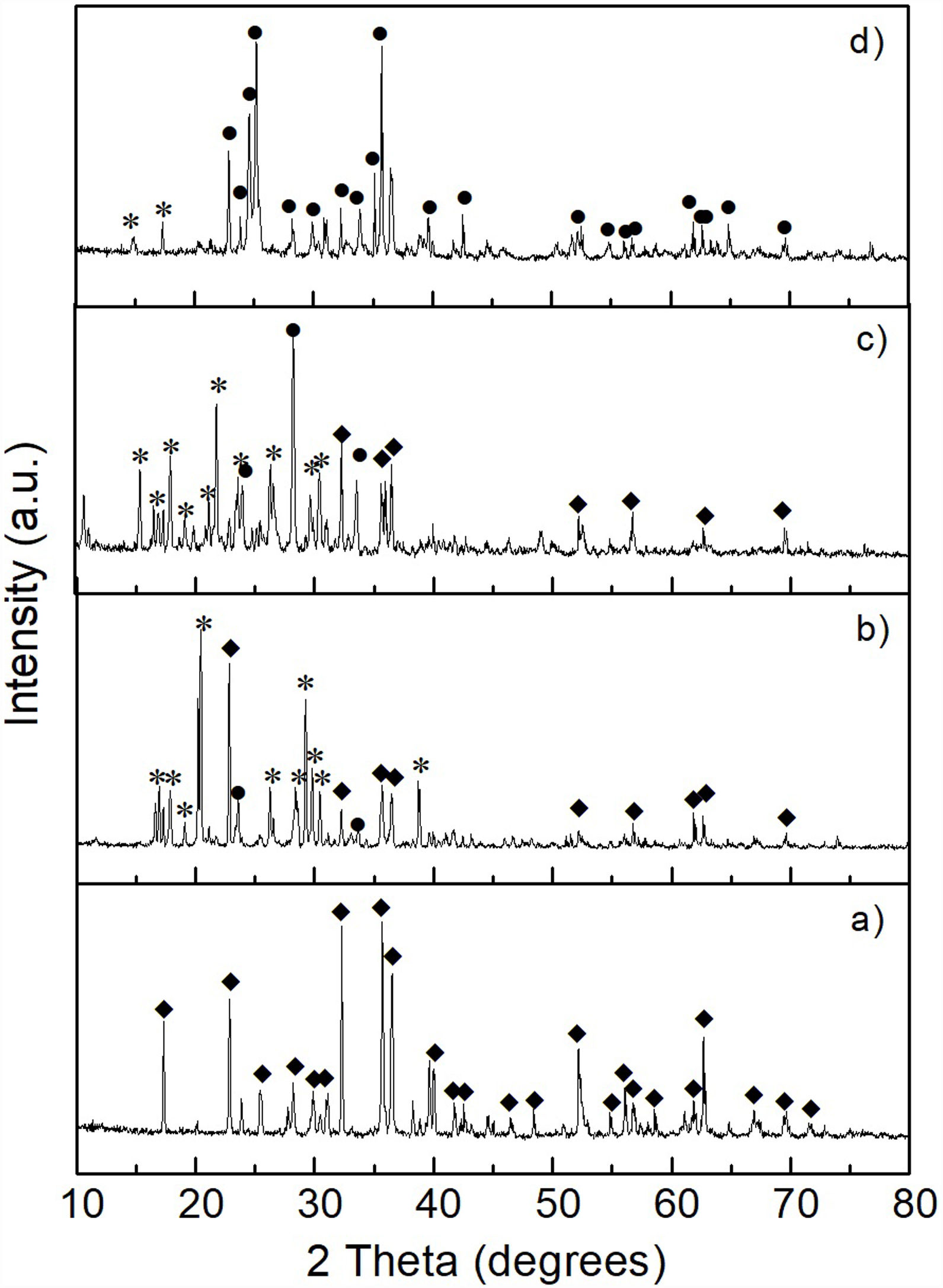
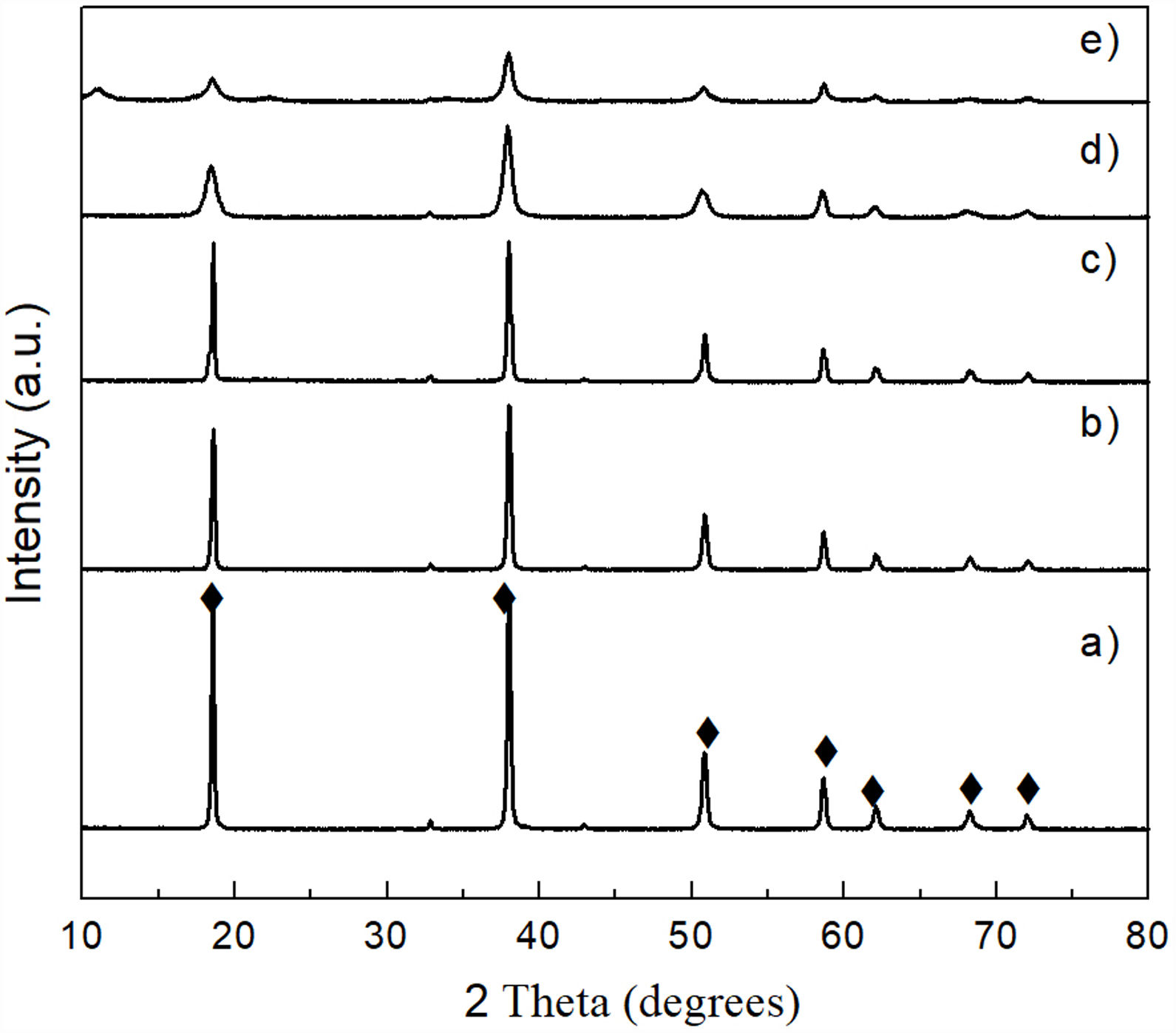
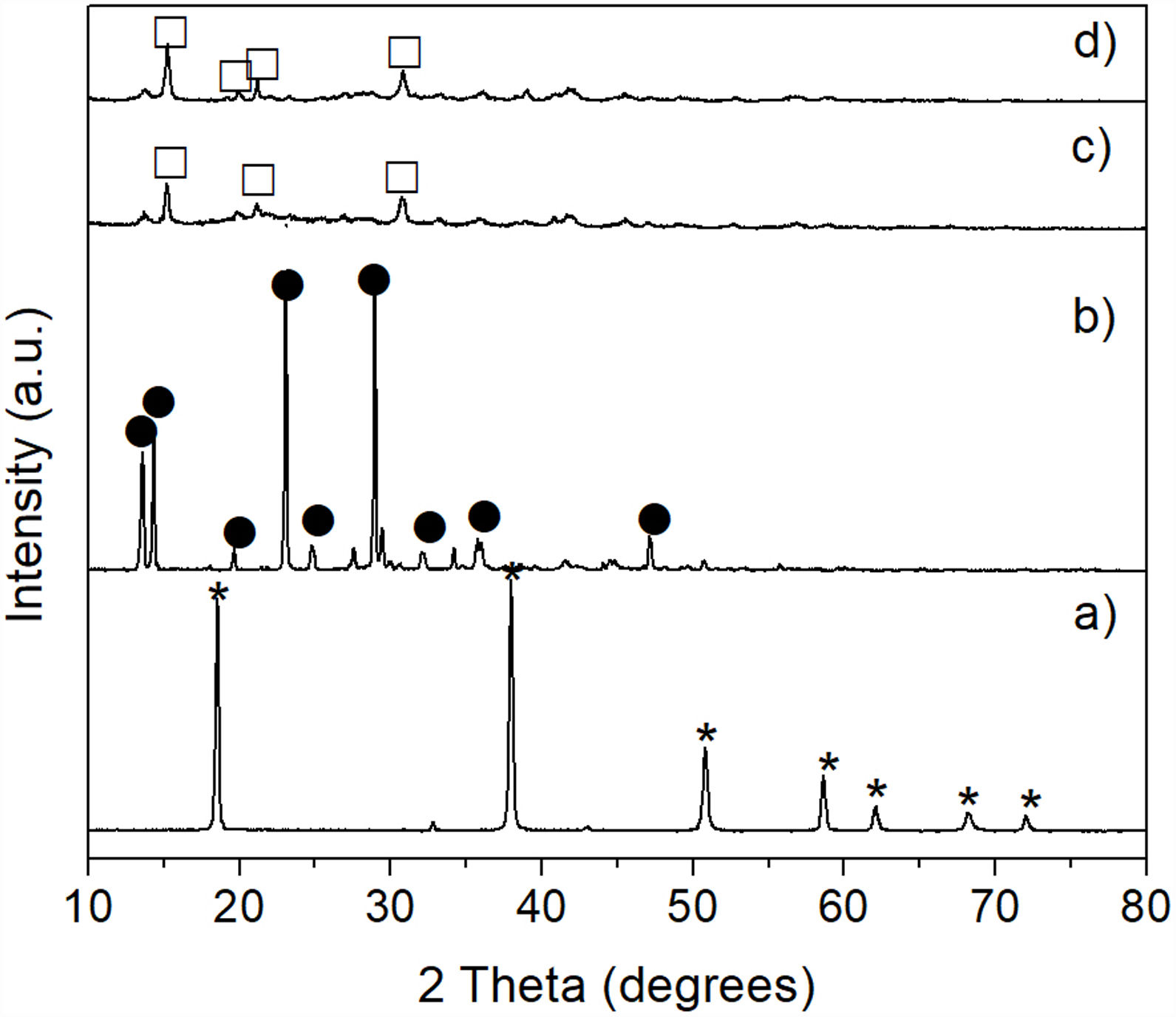
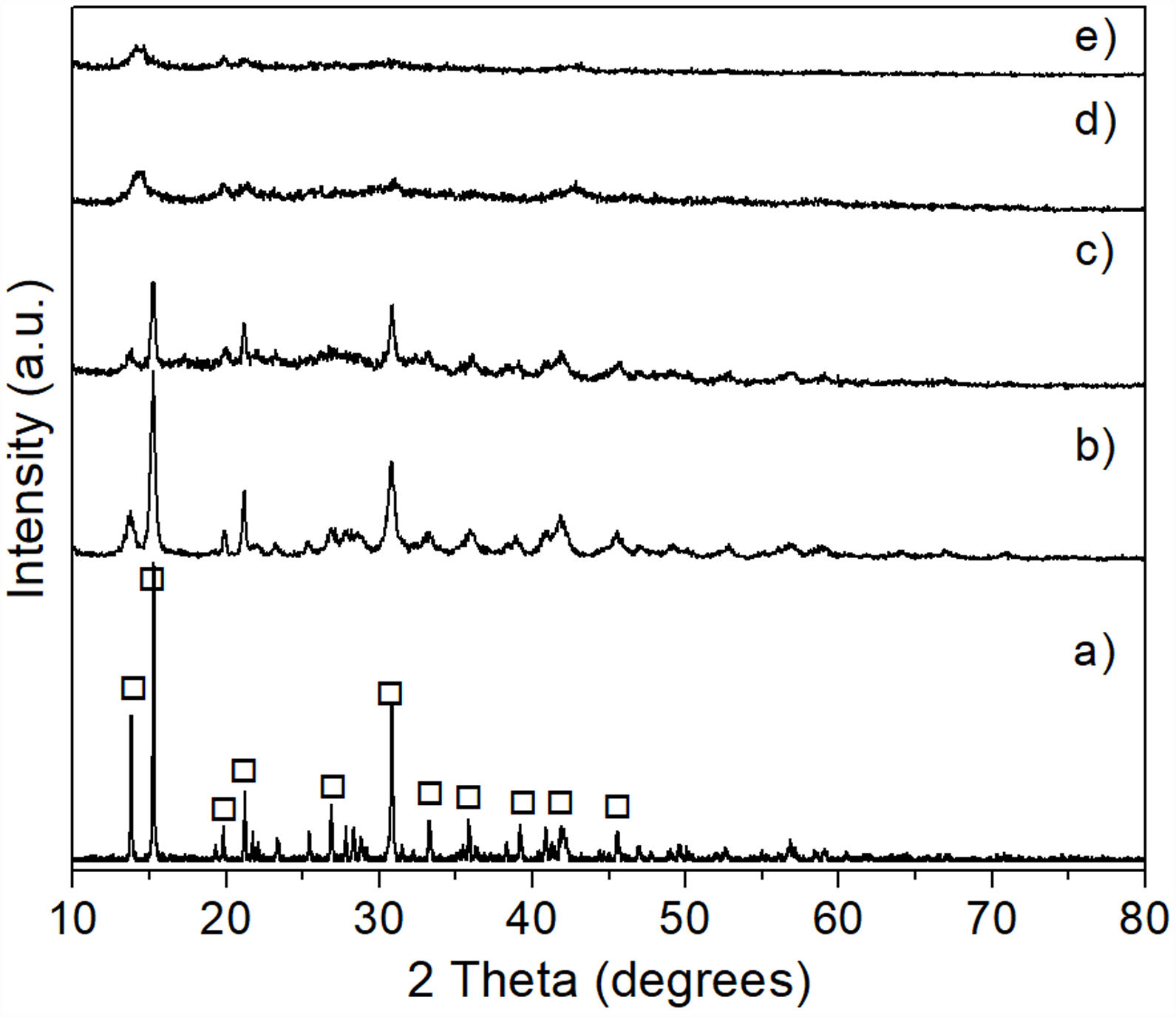
Results and discussionCharacterization of reacted olivine-(NH4)2SO4 mixturesThe XRD pattern obtained after carrying out the solid state reaction of the olivine mineral with ammonium sulfate at 300°C/1h (Fig. 3b), showed the presence of a considerable residual amount of both materials, as well as the presence of a small amount of MgSO4 formed. Upon increasing the temperature to 400°C/1h (Fig. 3c) a slight decrease in the residual amount of the starting materials was observed, as well as a slight increase in the amount of MgSO4 formed. In contrast, at 500°C/1h (Fig. 3d) only the presence of a relatively small amount of unreacted (NH4)2SO4 was detected, together with the almost complete disappearance of the characteristic peaks of olivine, with a predominance of MgSO4 in the sample. Probably the amount of FeSO4 and SiO2 formed in this sample was relatively small, in addition to the second phase being present in an amorphous state, which is why neither of them was detected in the XRD pattern.
, and after reaction of olivine with (NH4)2SO4 at 300°C (b), 400°C (c), and 500°C (d) for 1h. Key: ♦ Olivine, • MgSO4, and * (NH4)2SO4.")
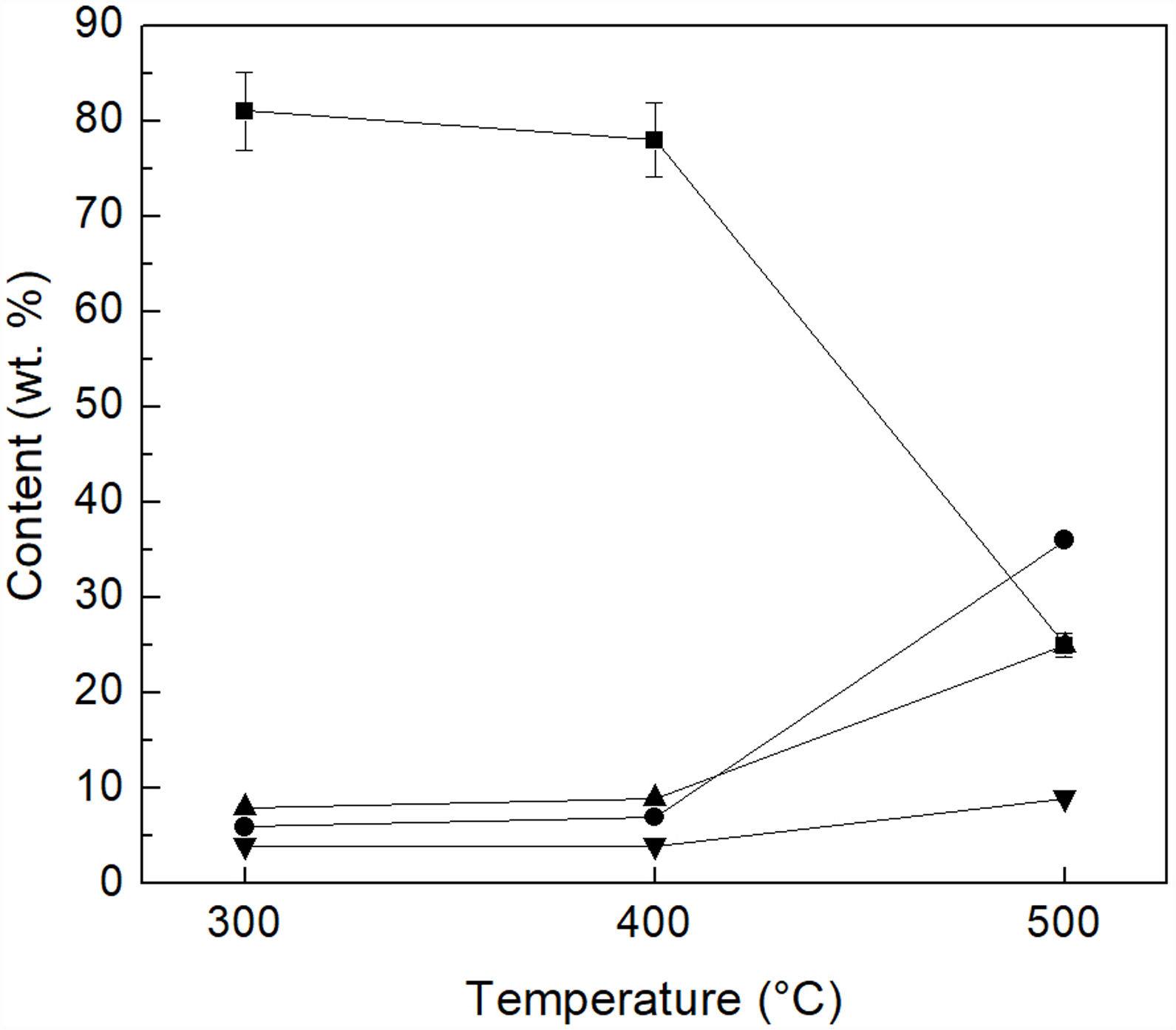
In order to confirm these results, a sample of each of the heat-treated materials was mixed with deionized water. The reaction products (MgSO4, FeSO4 and SiO2) and the residual unreacted (NH4)2SO4 were dissolved in the water, and the solid (basically unreacted olivine) was filtered off. Then, the solution was analyzed by ICP, and the amount of residual (NH4)2SO4 present in the heat-treated powders, as well as the amount of Mg, Fe and Si that was extracted from olivine, were estimated from the results obtained. Fig. 4 reports the percentage of unreacted (NH4)2SO4, with respect to the stoichiometric amount required according to reaction 1. The percentage of Mg, Fe and Si that was extracted from olivine is also reported in the same figure, with respect to the content of these elements in the initial mineral. As can be seen, 81% and 78% of unreacted (NH4)2SO4 were obtained at 300°C/1h and 400°C/1h, respectively, while the corresponding percentages extracted from the olivine were 6% and 7% for Mg, and 8% and 9% for Si, in addition to 4% of Fe for both treatment temperatures. However, the increase in the reaction temperature to 500°C/1h markedly favored the extraction of Mg, Si and Fe from olivine, by which it was possible to extract 36% of Mg, 26% of Fe and 9% of Si, from the sample treated at this temperature. Consequently, under these conditions the percentage of unreacted (NH4)2SO4 decreased sharply to 25%.
, Si () and Fe () in the aqueous solution, as well as of unreacted (NH4)2SO4 remaining in the samples after reaction with (NH4)2SO4 for 1h ().")



It is known [21] that (NH4)2SO4 decomposes at 400°C into NH3, SO2, H2O and N2, with the formation of (NH4)2S2O7 as a transitory phase, according to consecutive reactions 3 and 4:
This suggests that in the temperature range from 300 to 400°C the reaction between olivine and (NH4)2SO4 can be confidently represented by reaction 1. However, although in this work MgSO4 formation continued in the temperature range from 400 to 500°C, the reaction mechanism most likely changed due to the gradual thermal decomposition underwent by (NH4)2SO4. Nduagu et al. [25] proposed something similar for the case of the solid-state reaction between serpentine [Mg3Si2O5(OH)4] and (NH4)2SO4. Therefore, the steep decrease that occurs in the amount of the latter compound in the samples as the treatment temperature increases from 400 to 500°C can be explained partly by its reaction with olivine, and partly by the losses due to its thermal decomposition, forming gaseous products.
In terms of the reaction extent, the best results were obtained for the mixture of olivine ore with (NH4)2SO4 reacted at 500°C/1h. For this reason, from now on all reported results will correspond to mixtures reacted under such conditions.
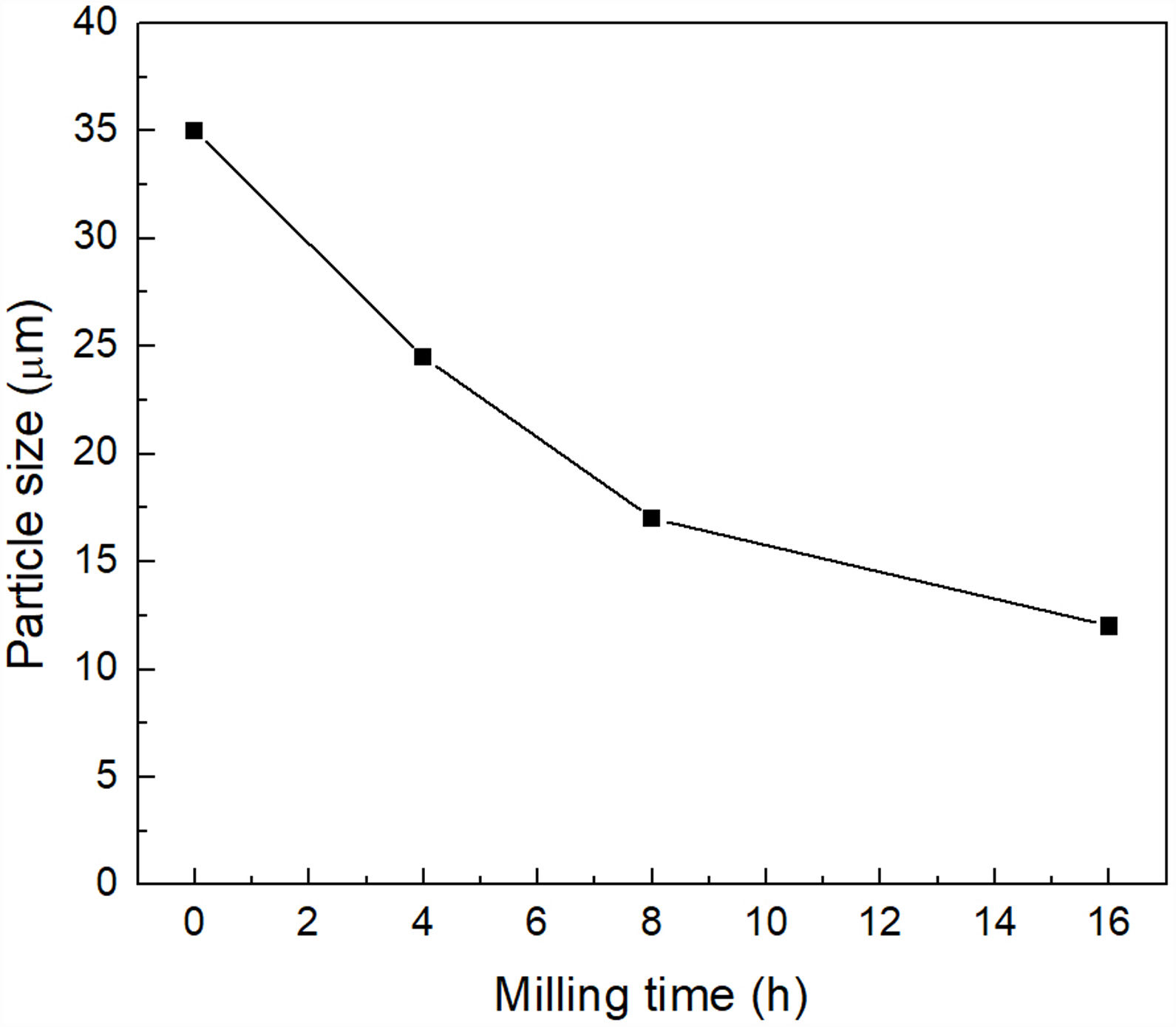
Characterization of mechanically activated materialsThe mean particle size of the material reacted at 500°C/1h and then mechanically activated for different times is presented in Fig. 5. It can be seen that the largest change occurs during the first eight hours of milling, after which the mean particle size practically no longer decreases, reaching a value of ∼13μm after 16h of mechanical activation.
 obtained as a function of milling time (mixture treated at 500°C/1h). The experimental points are joined by a line only for visualization purposes.")
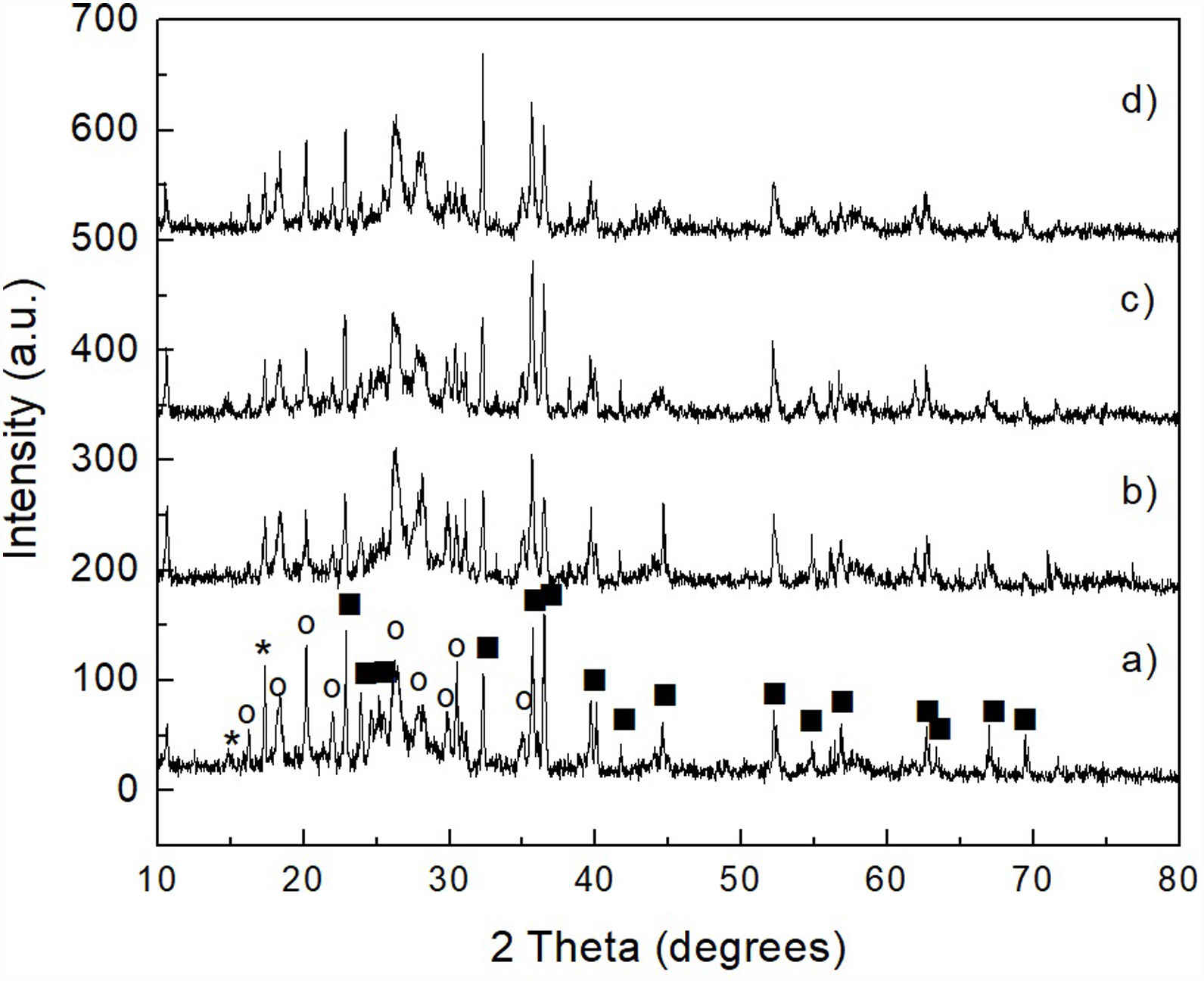
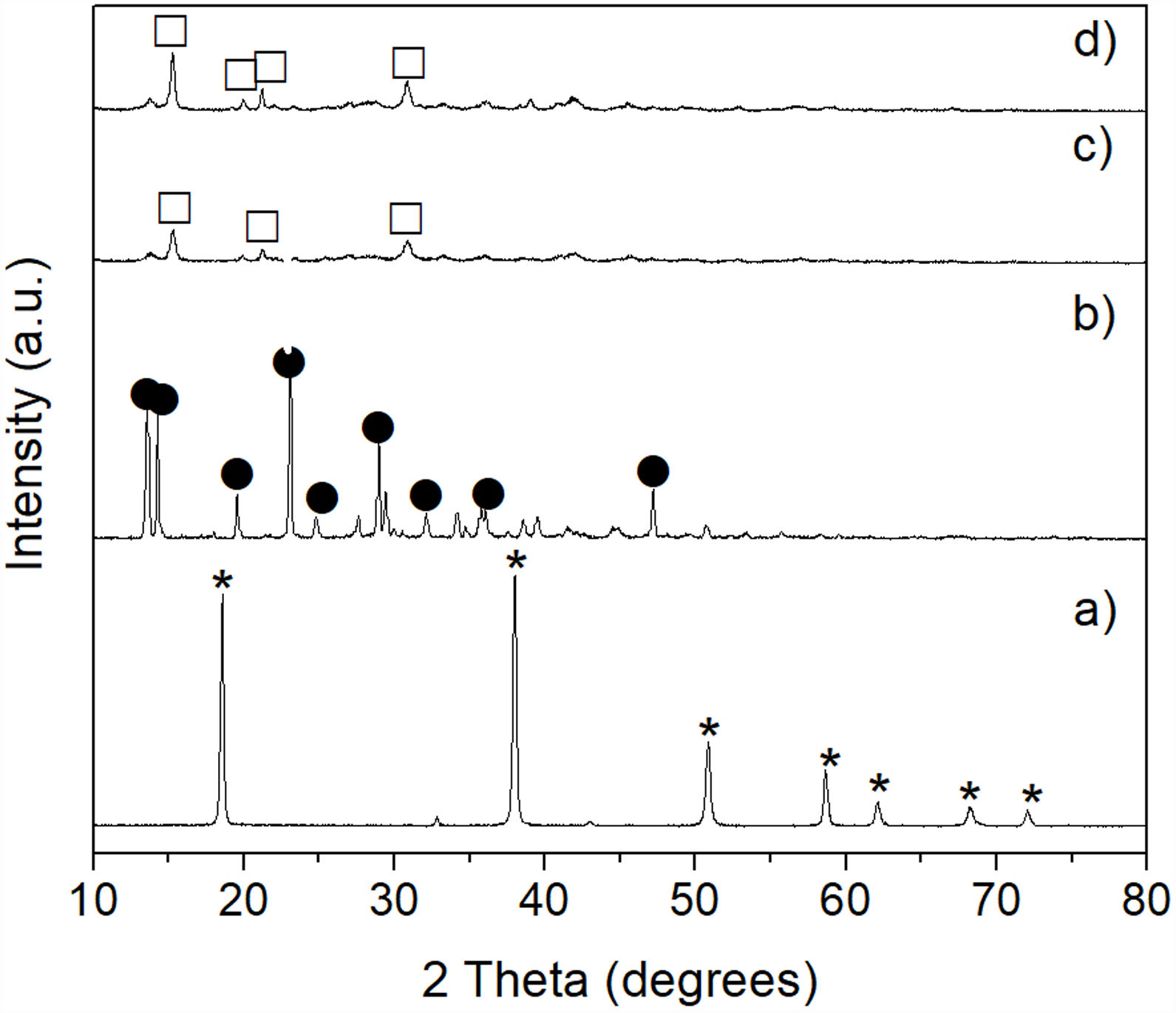
The results obtained by XRD are shown in Fig. 6, where the presence of the same phases [(Mg,Fe)2SiO4, MgSO4 and (NH4)2SO4] can be observed in all cases, regardless of the milling time. Again, probably in these samples the presence of crystalline FeSO4 and SiO2 was not detected due to the small amount of both phases, together with the likely amorphous state of the second one. This is confirmed by the amorphous halo located approximately in the range 2θ between ∼20° and ∼35° in the samples milled for 4–16h (Fig. 6b–d, respectively), which agrees with other literature reports (see, for instance, Refs. [26,27]). Another change that can be seen in the XRD patterns is a slight decrease in the intensity of the peaks with the increase in milling time, especially for magnesium and ammonium sulfates, which is particularly accentuated in the case of this last compound. This is more evident after 16h of mechanical activation (Fig. 6d). This behavior can be attributed to the fine particle size achieved after milling, as well as to the amorphization that these phases undergo when subjected to a prolonged milling process. In the particular case of (NH4)2SO4, however, one more possibility is that this compound undergoes a gradual decomposition during milling, which would reduce its amount in the material with the increase in the duration of the mechanical activation. This would not be rare considering the relatively low decomposition temperature of (NH4)2SO4. This would cause the amount of this compound to decrease with an increment in milling time. Analyzing reaction 2, according to Le Châtelier's principle, it is clear that the more (NH4)2SO4 is dissolved into the aqueous solution, the precipitation of brucite will be less favorable. Therefore, the more remnant (NH4)2SO4 is decomposed during mechanical activation, there will be a smaller amount of it that can dissolve in water, and the precipitation of brucite will be more favorable. This explains why a greater amount of precipitate is obtained by increasing the milling time, as will be discussed in greater depth later.
Removal of Fe and Si from the mechanically activated materials, and mechanically activated for 4h (b), 8h (c) and 16h (d). Key: MgSO4, (Mg,Fe)2SiO4, and (NH4)2SO4.")


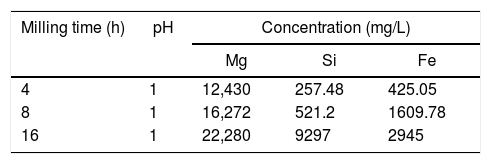
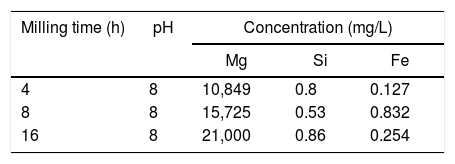
Tables 1 and 2 show the results of ICP obtained before and after the extraction of Fe and Si from the solution resulting from the dissolution in water of the products of reaction 1 [MgSO4, FeSO4 and SiO2], together with the dissolution of the remnant unreacted (NH4)2SO4.
In Table 1 it can be seen that the concentration of the three analyzed elements (Mg, Si and Fe) increases in the solution as the milling time increases, which indicates that the mechanical activation promotes their recovery from the olivine mineral, as expected. The amount of Mg extracted after 16h of milling was ∼80% greater than that extracted with 8h of mechanical activation. However, for the same increment in the milling time, the observed increase in the amount of Si and Fe extracted from the mineral was much greater. This indicates that by increasing the mechanical activation time it becomes increasingly important to efficiently remove both elements from the solution, prior to the precipitation of brucite from it. Fortunately, as can be seen in Table 2, after raising the pH of the solution from 1 to 8, the solution practically becomes free from Si and Fe. For all the milling times considered, the mentioned increase in the pH of the solution allows to remove more than 99.5% of the Si and Fe contained in it. On the other hand, under these conditions the concentration of Mg in the solution shows only slight variations, for any milling time, which indicates that the process of removal of Si and Fe from the solution does not cause a considerable loss of Mg from it.
Precipitation of brucite from the solutions free from Si and FeAfter the removal of Si and Fe from the solution, most of the MgSO4 produced by reaction 1 and all the unreacted (NH4)2SO4 remained dissolved in it. The addition of NH4OH to this solution provoked the precipitation of brucite, for all the milling times used.
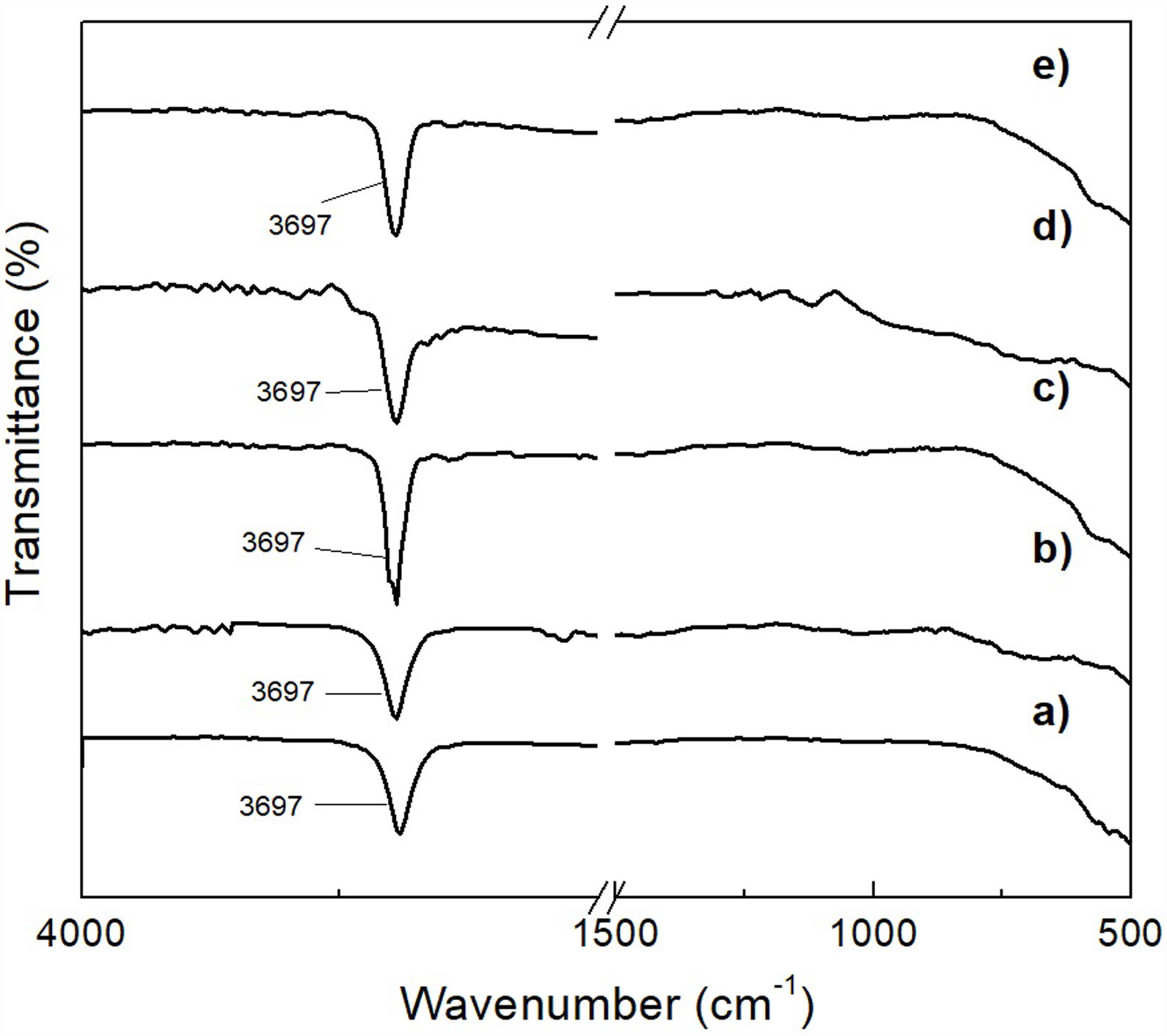
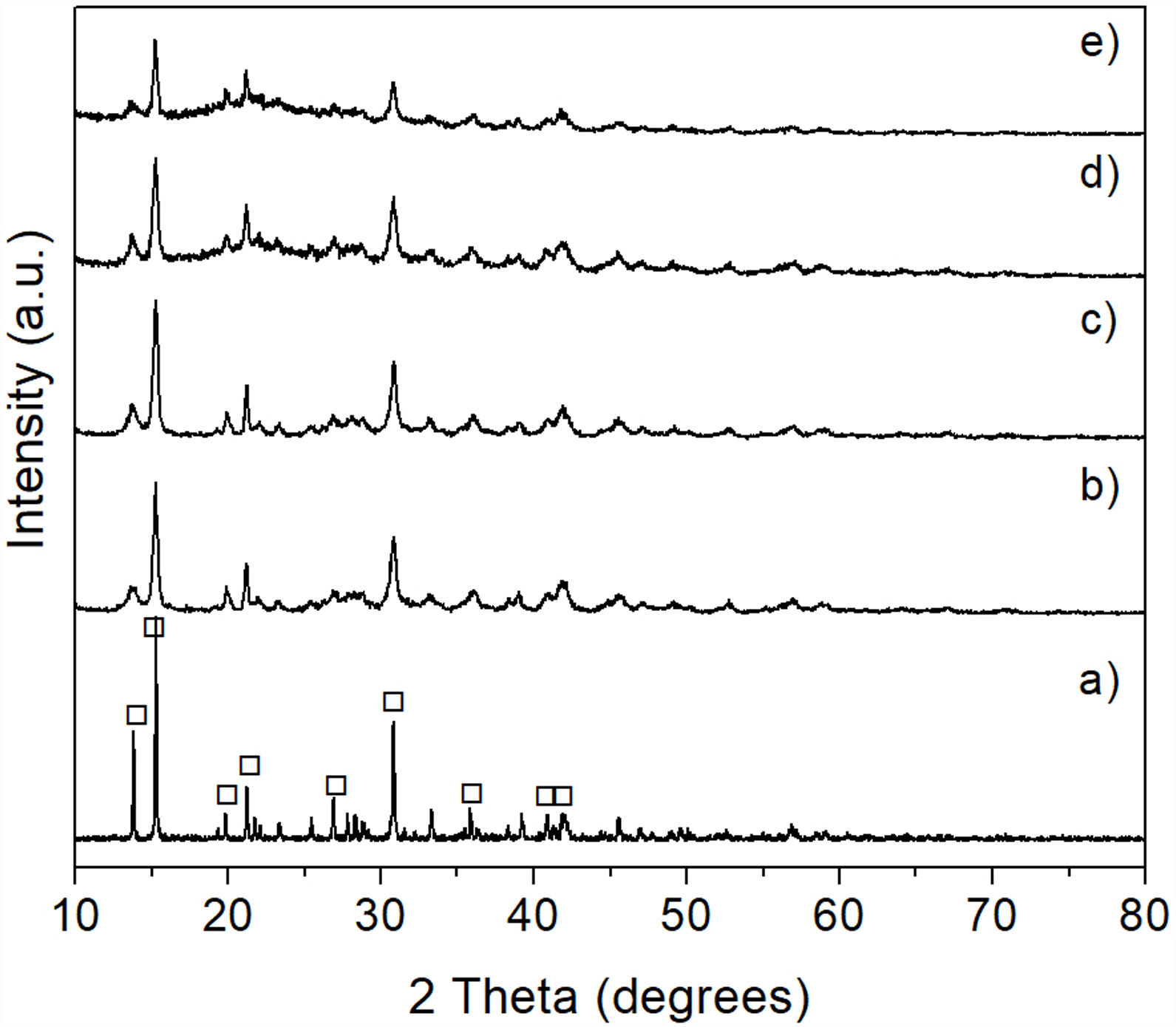
This was verified by XRD analysis, the results of which are presented in Fig. 7, where reagent grade brucite (JCPDS card no. 044-1482) was included for comparison purposes (Fig. 7a). As can be seen, in all cases only the presence of brucite was detected in the precipitate, with a clear decrease in the intensity of its reflections, together with a concomitant slight widening of them, with increasing milling time. This can be attributed to a stronger tendency toward the amorphization shown by the precipitate as the time of mechanical activation increases.
2 (JCPDS card no. 044-1482) (a) and of Mg(OH)2 obtained from powder mechanically activated for 0 (b), 4 (c), 8 (d) and 16h (e). Key: ♦ Mg(OH)2.")
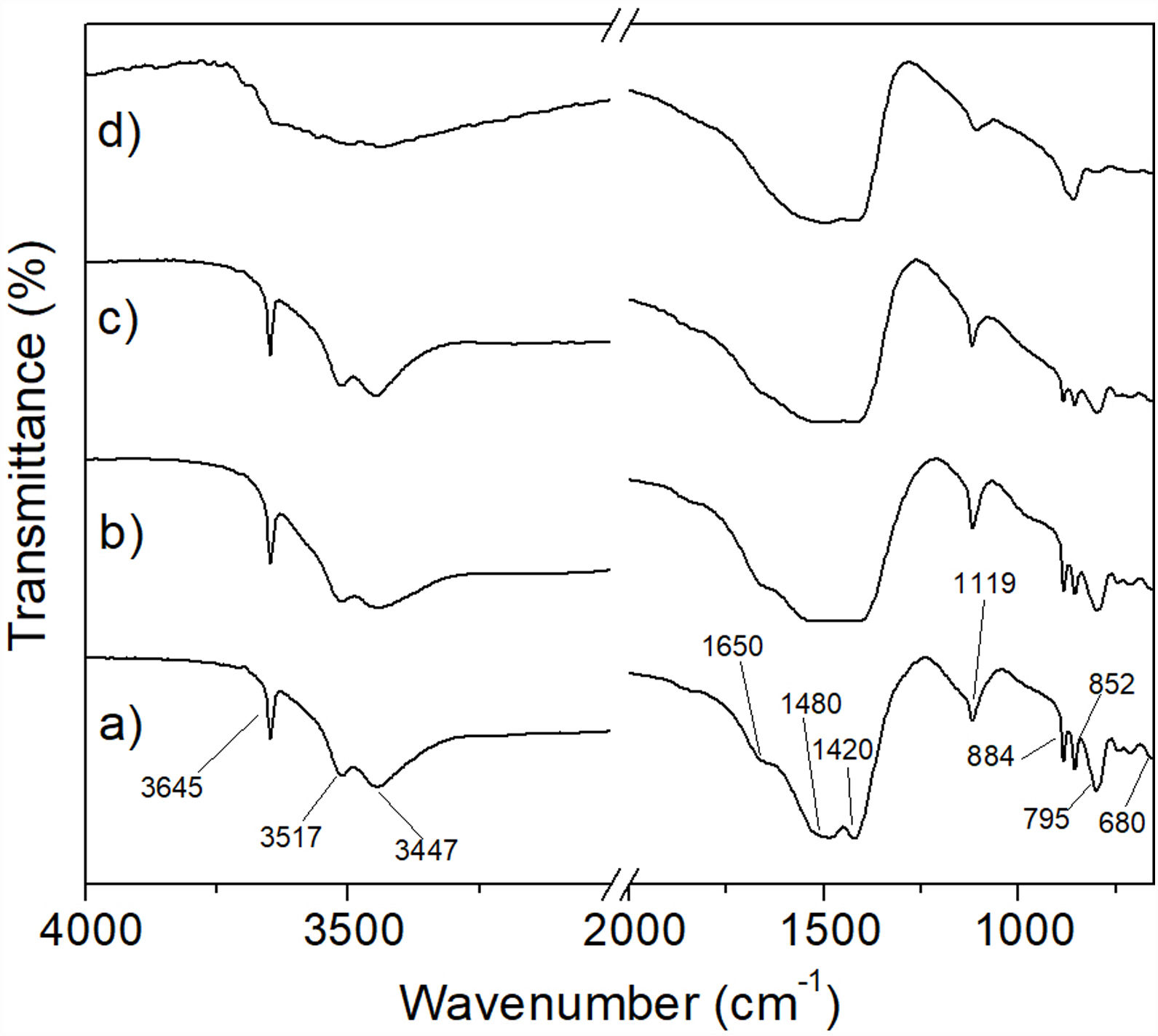
This was supported by the results shown in Fig. 8, which presents the FT-IR spectra obtained for the precipitates, as well as for reagent grade brucite, for comparison purposes. In these spectra, the presence of a strong absorption band located at a wavelength of 3697cm−1 can be highlighted, which corresponds to asymmetric stretching vibrations of the functional groups –OH of brucite [28]. Thus, this study confirmed that the formation of brucite took place in all cases.
2 (a) and of Mg(OH)2 obtained from powder mechanically activated for 0 (b), 4 (c), 8 (d) and 16h (e).")
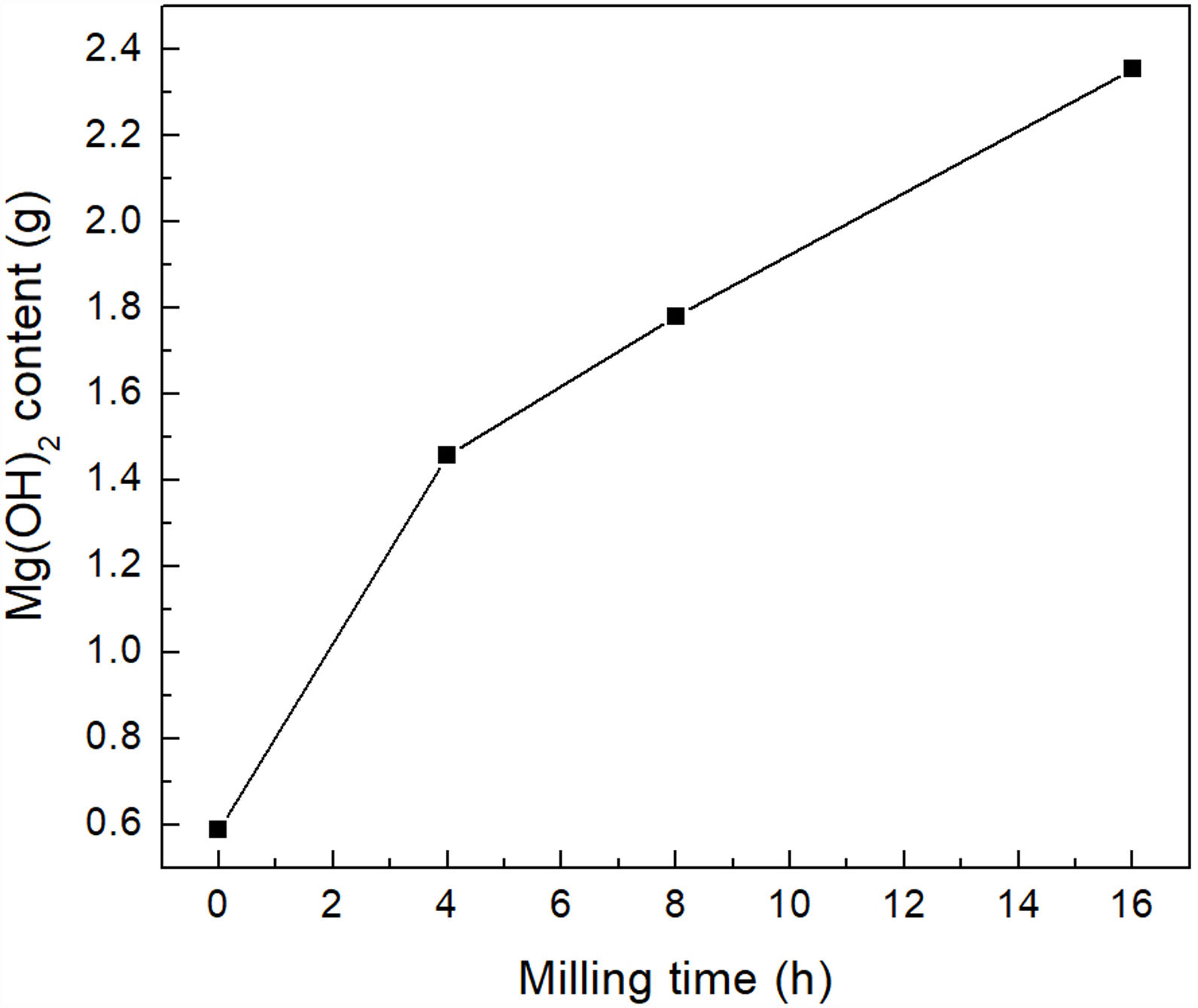
The main benefit of the mechanical activation was reflected in the amount of brucite obtained after the last processing stage. According to the analysis performed, for each 5g of powder, without mechanical activation it was possible to obtain only 0.500g of brucite, while with 4h of milling it was possible to obtain 1.458g of the precipitate, which represents an increase of about 200%. On the other hand, with 8 and 16h of milling, 1.781 and 2.355g of brucite were obtained, respectively, which represents an increase of ∼260% and ∼370%, respectively, with respect to the material without mechanical activation. Therefore, the amount of precipitated brucite clearly increased with increasing milling time, see Fig. 9.
CO2 absorption by the precipitated brucite2 obtained by precipitation as a function of milling time.")
To carry out these tests, the brucite obtained from the powder mechanically activated for 16h was used. Fig. 10 shows the XRD patterns obtained after CO2 absorption tests carried out at 50, 100 and 150°C for 24h for brucite suspended in H2O, under a continuous flow of CO2. The X-ray diffractogram of reagent grade brucite is shown in Fig. 10a, for comparison purposes. In Fig. 10b a notable change is observed at 50°C, due to the formation of nesquehonite (MgCO3·3H2O), which is a hydrated magnesium carbonate. It should be emphasized that in the XRD pattern the presence of residual brucite was not appreciated, so that this compound was completely transformed into nesquehonite at this temperature. It was deduced, based on our results as well as on literature reports [29], that in this work the formation of nesquehonite could have occurred according to reaction 5:
2 (JCPDS card no. 044-1482) (a), and of Mg(OH)2 after CO2 absorption at 50°C (b), 100°C (c), and 150°C for 24h. Key: * Mg(OH)2, • MgCO3·(H2O)3, and Mg5(CO3)4(OH)2·4H2O.")

When the reaction temperature was increased either to 100 or 150°C a new phase appeared, which corresponded to hydromagnesite [Mg5(CO3)4(OH)2·4H2O] (Fig. 10c and d). This was due to a partial dehydration of nesquehonite, as well as to the loss of part of its CO2 content. This transformation is consistent with that reported by Davies and Bubela [30], who mentioned that when nesquehonite is submerged in distilled water and heated to 52°C, it is easily transformed into hydromagnesite through the formation of an intermediate phase with an approximate composition of MgCO3·2H2O, which is accompanied by a great loss of water. Therefore, in our case the observed nesquehonite-to-hydromagnesite transformation could have occurred according to reactions (6) and (7):
A similar behavior was observed in the CO2 absorption tests carried out for a period of 72h. At low temperature (50°C), the formation of nesquehonite was also obtained, which was subsequently transformed into hydromagnesite by raising the temperature either to 100 or 150°C (Fig. 11).
2 (JCPDS card no. 044-1482) (a), and of Mg(OH)2 after CO2 absorption at 50°C (b), 100°C (c), and 150°C for 72h. Key: * Mg(OH)2, • MgCO3·3H2O, and Mg5(CO3)4(OH)2·4H2O.")
Davies and Bubela [30] observed that after 72h at 52°C nesquehonite only partially transformed, forming a small amount of hydromagnesite plus the mentioned intermediate phase. The total disappearance of nesquehonite occurred after one week of treatment at that temperature, and only hydromagnesite and the intermediate phase were detected in the treated material. In this work, the mechanical activation produced significantly different results in several aspects with respect to those reported by the mentioned authors. First, the fact that the intermediate phase was not detected at any of the temperatures or treatment times used by us indicated that in this work nesquehonite was able to transform into hydromagnesite at a rate considerably faster than that observed by Davies and Bubela [30]. In our case, 24h of treatment at 100°C were enough for complete transformation of nesquehonite into hydromagnesite, which was not achieved by the mentioned researchers even after a week, as already mentioned. This could have important economic repercussions for those who wish to obtain high purity hydromagnesite from brucite or nesquehonite, since our method is relatively quick and efficient and only requires a relatively low reaction temperature.
Finally, in terms of CO2 capture, these results indicate that it should be preferred to carry out the CO2 absorption tests at 50°C/24h with brucite suspended in water, in which nesquehonite is formed. This corresponds to the largest CO2 capture achieved in this work, since the formation of hydromagnesite from nesquehonite at higher temperatures with the same treatment time, implies the release of some of the previously captured CO2. The nesquehonite resulting from the process employed in the present work could be used as the basis for the manufacture of various materials useful for the construction industry. See for instance the work by Glasser et al. [31]. However, it is worth mentioning that one of the main drawbacks that a practical industrial application would have is that the reaction times would have to be relatively long (minimum 24h), since the reaction kinetics is relatively slow under the conditions studied.
CO2 absorption by hydromagnesiteTrying to achieve a maximum capacity for CO2 capture, CO2 absorption tests were carried out using the obtained hydromagnesite, for which the experimental arrangement shown in Fig. 2 was employed. It was intended to promote the transformation of hydromagnesite into magnesite (anhydrous magnesium carbonate).
The results of the analyzes performed by XRD for the materials produced in these tests are shown in Fig. 12, where it can be seen that the increase in the reaction temperature from 50 to 100, 150 or 200°C produces a gradual amorphization of the material, which is more noticeable at the two higher temperatures. This behavior could be attributed to the formation of amorphous magnesite. For instance, Dell and Weller [32] heated hydromagnesite in a stream of nitrogen or carbon dioxide, and with both gases a great loss of water was observed in the temperature range of 300–350°C, which was accompanied by the formation of an amorphous magnesium carbonate containing approximately 0.5 moles of water per mole of magnesia. In air, a small loss of CO2 started at 400°C, while in a CO2 atmosphere this process started at ∼430°C. Subsequently, recrystallization of magnesite was observed to occur at 510°C with both gases, which subsequently decomposed into magnesia at ∼560°C in air, and at ∼590°C in CO2. Therefore, in the presence of a CO2 atmosphere, the release of this gas from magnesium carbonates was delayed at high temperatures, which has been observed many times in the past.
 (a), and of hydromagnesite after CO2 absorption at 50°C (b), 100°C (c), 150°C, (d) and 200°C (d) for 3h. Key: Mg5(CO3)4(OH)2·4H2O.")
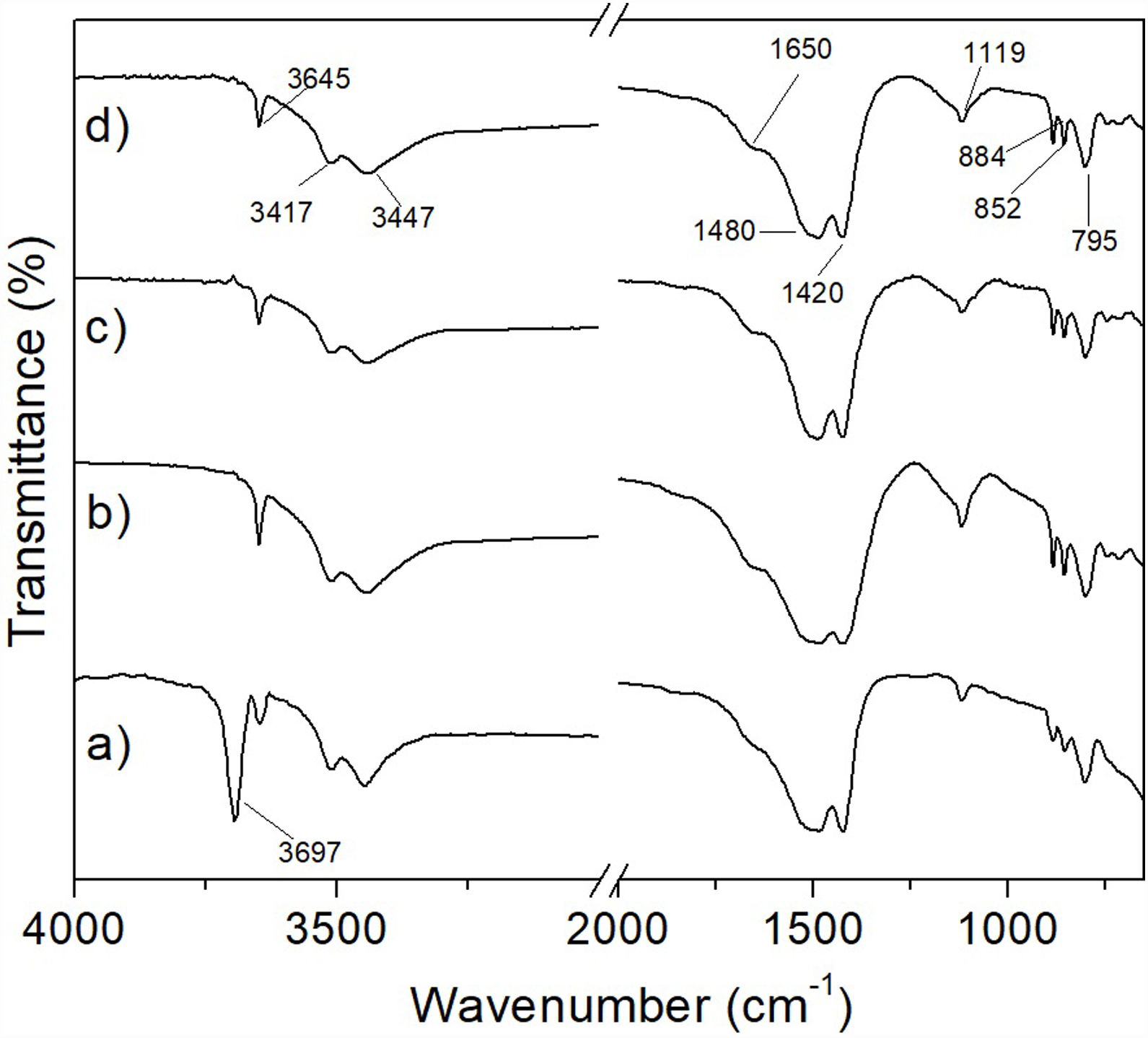
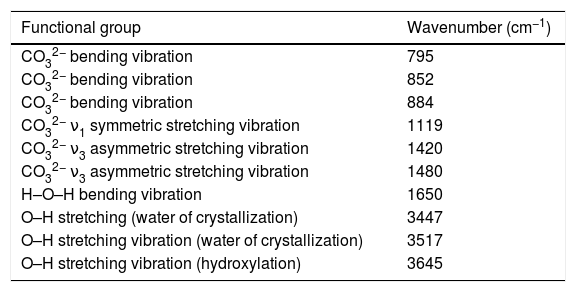
In this work, although it was not possible to observe in a clear way the formation of MgCO3 by means of the XRD technique, the amorphization underwent by the material suggested that it was likely that the mentioned phase was formed in it. In order to verify this, an FT-IR analysis was carried out for hydromagnesite subjected to CO2 absorption tests at 50, 100, 150 and 200°C for 3h. The results are presented in Fig. 13. Table 3 shows the assignment given to the absorption bands observed in the FT-IR spectra of the previous figure. The assignment of the observed absorption bands was made in accordance with Botha and Strydom [33]. As can be seen, in all cases the observed absorption bands can be attributed to functional groups present in hydromagnesite.
, 100°C (b), 150°C (c), and 200°C (d) for 3h.")
Assignment of the absorption bands found in the FT-IR spectra obtained for hydromagnesite.
| Functional group | Wavenumber (cm−1) |
|---|---|
| CO32− bending vibration | 795 |
| CO32− bending vibration | 852 |
| CO32− bending vibration | 884 |
| CO32− ν1 symmetric stretching vibration | 1119 |
| CO32− ν3 asymmetric stretching vibration | 1420 |
| CO32− ν3 asymmetric stretching vibration | 1480 |
| H–O–H bending vibration | 1650 |
| O–H stretching (water of crystallization) | 3447 |
| O–H stretching vibration (water of crystallization) | 3517 |
| O–H stretching vibration (hydroxylation) | 3645 |
The results of XRD obtained after subjecting the hydromagnesite to the CO2 absorption tests at 50, 100, 150 or 200°C for 6h are shown in Fig. 14. The XRD pattern obtained for reagent grade hydromagnesite is shown in Fig. 14a, for comparison purposes. The X-ray diffractogram of Fig. 14b, corresponding to 50°C, clearly shows that the transformation of the material to the desired phase was not achieved at this temperature. However, when the temperature was increased either to 100 or 150°C, a considerable diminution in the intensity of the characteristic reflections of hydromagnesite was observed (Fig. 14c and d). This was more pronounced at 150°C, until the crystallinity of the sample was almost fully lost when a temperature of 200°C was reached (Fig. 14e). As mentioned earlier, this behavior can be attributed to the formation of amorphous MgCO3 from the hydromagnesite, with an increment in the temperature of the tests.
 (a), and of hydromagnesite after CO2 absorption at 50°C (b), 100°C (c), 150°C (d), and 200°C (e) for 6h. Key: Mg5(CO3)4(OH)2·4H2O.")
To corroborate the previous results, analyzes were carried out using the FT-IR technique. In Fig. 15 it can be seen that the spectra obtained for the materials subjected to the CO2 absorption tests in a temperature range from 50 to 150°C for 6h (Fig. 15a–c), correspond to hydromagnesite (see Table 3). In contrast, the spectrum of the sample treated at 200°C/6h, Fig. 15d, shows the following significant changes with respect to the spectra obtained for lower treatment temperatures: (1) the absorption bands located at 3645, 3517 and 3447cm−1, the first one attributed to physically bound water, and the last two attributed to water of crystallization, are replaced by a single broad band, which indicates that the material still has an appreciable amount of hydration water; (2) the absorption bands located at 1420, 1480 and 1650cm−1, the first two attributed to CO32− ν3 asymmetric stretching vibrations, and the third to H-O-H bending vibration, merge into a single intense and wide band, centered approximately at 1450cm−1; (3) the intensity of the band located at 1119cm−1, attributed to CO32− ν1 symmetric stretching vibration, decreases dramatically; (4) the bands located at 795, 852 and 884cm−1, attributed to CO32− bending vibrations, disappear, and (5) a new small band located at 856cm−1 appears. All these changes make the FT-IR spectrum corresponding to 200°C/6h significantly different from those obtained at lower temperatures. This suggests that under these conditions hydromagnesite almost completely disappeared, being largely replaced by hydrated magnesite.
, 100°C (b), 150°C (c), and 200°C (d) for 6h.")
This is supported by the fact that the most important absorption bands observed in Fig. 15d coincide with some of the most representative bands reported by Gopinath and Gunasekaran for magnesite [34]. These authors observed the following absorption bands in the FT-IR spectrum of magnesite: (a) a very strong broad band located at 1446cm−1, which can be assigned to asymmetric C-O stretching vibrations, (b) an intense band located at 856cm−1, which is characteristic of the CO3 asymmetric deformation, (c) two weak bands located at 1080cm−1 and 680cm−1 that could be assigned to the C-O and CO3 symmetric deformation vibrations, respectively, (d) four bands located at 1446cm−1, 856cm−1, 1080cm−1 and 680cm−1, which correspond to the fundamental modes of CO3 ion present in MgCO3, and (e) a strong band located near 887cm−1, due to the Mg-O stretching vibration.
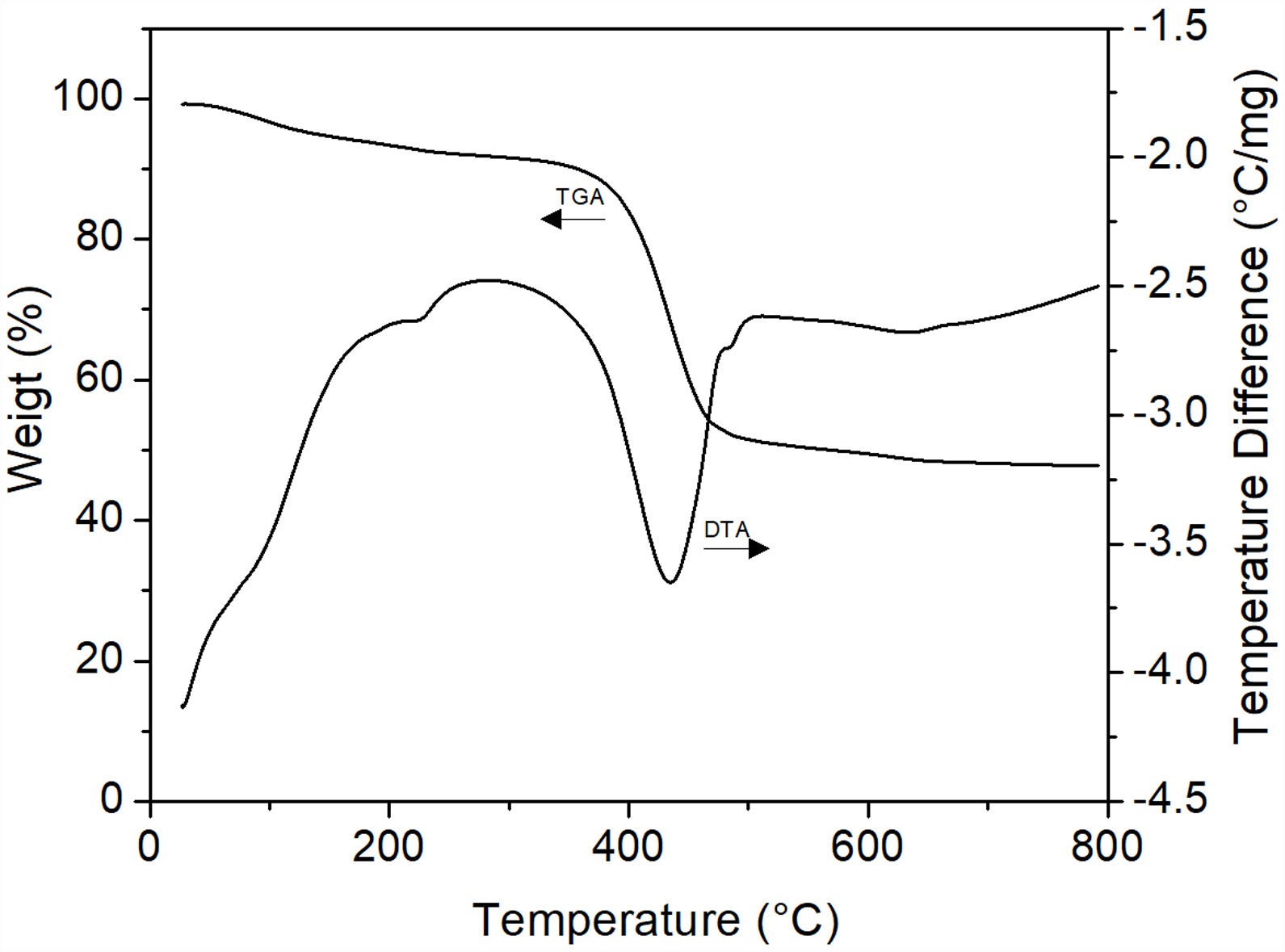
The samples that presented the largest amorphization degree (those treated at 200°C/6h, Fig. 14e) were analyzed by DTA/TGA in order to confirm the formation of MgCO3 in them. The results of these analyzes are shown in Fig. 16, where it can be seen that the stage of greatest weight loss (∼42%) occurs in the temperature range of 380–450°C, which is attributed to the decarbonation of the sample. Considering that the theoretical weight loss to obtain MgO from MgCO3 and hydromagnesite is 52.2% and 69% (38% CO2 and 31% H2O), respectively, it follows that probably what was obtained under the employed conditions was basically amorphous MgCO3 containing an appreciable amount of H2O. However, based on the results of XRD (Fig. 14e), the presence in the material of a very small amount of remnant hydromagnesite was not ruled out. This is supported by the fact that between room temperature and ∼250°C there was a weight loss of ∼10%, corresponding to hydration water. This weight loss was suffered mainly by the amorphous phase, but it is known [33] that hydromagnesite also undergoes a weight loss at the same temperature range. The presence of other stages of weight loss at other temperature ranges was not evident, so that the existence of small amounts of other magnesium carbonates could be confidently ruled out. Finally, the pronounced endothermic peak observed at ∼430°C in the DTA curve can be attributed to magnesite decomposition into MgO and CO2. It is important to note that the recrystallization of magnesite observed by Dell and Weller [32] at 510°C was not detected in this work, which could be another effect of mechanical activation.
Conclusions
The largest reaction extent was achieved for the olivine-(NH4)2SO4 mixture reacted in the solid state at 500°C/1h. These reaction conditions markedly favored the extraction of Mg, Si and Fe from olivine, when compared with that obtained using lower reaction temperatures.
When the material reacted under such conditions was mechanically activated, both MgSO4 and (NH4)2SO4 present in it became increasingly amorphous with increasing milling time, which was more accentuated for the case of the latter phase. It was hypothesized that the unreacted (NH4)2SO4 could also undergo a gradual partial decomposition into various gaseous products, with increasing time of milling.
Independently of milling time, it was possible to remove more than 99.5% of the Si and Fe contained in the solution resulting from the dissolution in water of the reaction products (MgSO4, FeSO4 and SiO2) and unreacted (NH4)2SO4 contained in the mechanically activated material, with a minimum loss of Mg.
It was possible to precipitate pure brucite from the aqueous solution free from Si and Fe. The amount of precipitated brucite increased with increasing milling time, which was attributed to the partial decomposition of the unreacted (NH4)2SO4 during mechanical activation, which negatively interferes with the precipitation process of brucite.
The CO2 absorption tests carried out using the precipitated brucite showed that after 24h at 50°C the latter compound was completely transformed into nesquehonite (MgCO3·3H2O). When temperature was increased to 100°C, with the same treatment time, nesquehonite was completely transformed into hydromagnesite [Mg5(CO3)4(OH)2·4H2O]. This was due to a partial dehydration of nesquehonite, as well as to the loss of part of its CO2 content occurring under such conditions. The CO2 absorption tests carried out using the obtained hydromagnesite, showed that at 200°C/6h it was almost completely replaced by amorphous hydrated magnesite.
Regarding the CO2 capture, the best results were obtained at 50°C/24h with brucite suspended in water, in which only nesquehonite was formed. The transformation of the latter compound into hydromagnesite at higher temperatures implied the release of some of the previously captured CO2.
The nesquehonite resulting from the CO2 capture process studied in this work could be used as the basis for the manufacture of various materials useful for the construction industry.
Author contribution statementC.M. García-Hernández made the experiments, characterized the initial and produced materials and wrote the draft paper.
J. López-Cuevas is the corresponding author, co-directed the experimental work, assisted in every aspect of the experimental and characterization work, and supervised the final manuscript.
C.A. Gutiérrez-Chavarría co-directed the experimental work, assisted in every aspect of the experimental and characterization work, and supervised the final manuscript.
Conflict of interest statementThe authors declare that they have no conflict of interest.
The authors greatly acknowledge Cinvestav-Saltillo for facilitating access and use of their facilities and experimental equipment to carry out this research. C.M.G.H. acknowledges CONACyT for granting her a Ph.D. scholarship.

