







A glass-ceramic biomaterial with near-eutectic composition of the pseudo-binary system diopside (CMS2, CaMgSi2O6)–tricalcium phosphate [C3P, Ca3(PO4)2], with a weight composition of 61% CMS2–39% C3P, was synthesized by the so-called “petrurgic method”. Three different cooling rates (0.5, 1 and 2°C/h) were used through the mushy zone. The synthesized materials showed a microstructure consisting of β-C3Pss (solid solution of CMS2 in C3P) primary skeletal dendrites, with a peculiar comb-like morphology, and which were embedded in a matrix of lamellar CMS2–β-C3Pss eutectic phase. The matrix also presumably contained a small amount of CaO–SiO2 glass segregated during formation of the β-C3Pss phase. The cooling rate used through the mushy zone affected the microstructure and phase composition of the synthesized materials. The dendrites of the primary β-C3Pss phase became smaller and thicker with decreasing cooling rate, while the level of substitution of Mg for Ca (x) decreased and that of Si for P (δ) increased in the latter phase, and the CMS2/β-C3Pss weight ratio tended to increase slightly in the interdendritic matrix, with increasing cooling rate.
Un biomaterial vitrocerámico con composición cercana a la eutéctica del sistema seudobinario diópsido (CMS2, CaMgSi2O6) - fosfato tricálcico [C3P, Ca3(PO4)2], con una composición en masa del 61% CMS2 - 39% C3P, fue sintetizado utilizando el llamado «método petrúrgico». Se usaron 3 diferentes velocidades de enfriamiento (0.5, 1 y 2°C/h) a través de la zona de transición sólido/líquido. Los materiales sintetizados mostraron una microestructura consistente en dendritas esqueléticas primarias de la fase β-C3Pss (solución sólida de CMS2 en C3P), con una morfología peculiar similar a un peine, embebidas en una matriz constituida por la fase eutéctica laminar CMS2 - β-C3Pss. La matriz probablemente también contenía una pequeña cantidad de vidrio CaO-SiO2 segregado durante la formación de la fase β-C3Pss. La velocidad de enfriamiento utilizada a través de la zona de transición sólido/líquido afectó la microestructura y la composición de fases de los materiales sintetizados. Las dendritas de la fase primaria β-C3Pss se volvieron más pequeñas y gruesas con una velocidad de enfriamiento decreciente, mientras que el nivel de sustitución de Mg por Ca (x) disminuyó y el de Si por P (δ) aumentó en la última fase, y la relación en peso CMS2/β-C3Pss tendió a aumentar ligeramente en la matriz interdendrítica, con una velocidad de enfriamiento creciente.
Osseointegration (ability to promote the growth of new bone) is one of the most important properties that biomedical implants must possess. To achieve this, the use of materials with an adequate structure of interconnected pores, similar to that of bone, has been proposed [1–4]. However, porous materials have poor mechanical properties. To solve this problem, dense ceramic materials of binary composition capable of generating in situ a suitable porous structure have been designed. In them, one of the phases dissolves into the physiological fluid, leaving behind a skeleton formed by the other phase, originating in this way a structure of interconnected pores. Subsequently, the phase that remains undissolved is transformed into hydroxyapatite [HAp, Ca10(PO4)6(OH)2], which is similar to the mineral phase of bone, by means of a pseudomorphic conversion process. Later, an adhesion interface is developed between the latter material and the bone tissue, through the formation of a HAp surface layer [5].
So-called Bioeutectic® is the most representative material showing the aforementioned behavior. It corresponds to the eutectic composition of the binary system wollastonite (CS, CaSiO3)–tricalcium phosphate [C3P, Ca3(PO4)2], which is constituted by 60% CS and 40% C3P (throughout this work, compositions are given in weight percent, unless otherwise specified), with a eutectic temperature of 1402±3°C. In this system, CS is the phase that dissolves and C3P is the phase that transforms pseudomorphically into HAp, in contact with the physiological fluids [5].
Another material with potential to exhibit a behavior similar to that of Bioeutectic®, although presumably with better mechanical properties, is the eutectic composition of the binary system diopside (CMS2, CaMgSi2O6)–C3P. This system was studied for the first time by Sata [6], who determined that there was a eutectic point located at 61% CMS2–39% C3P, at a temperature of 1311±4°C. This researcher mentioned that although this system can be described using a binary diagram, it is actually approximately binary, since evidence of the formation of a solid solution of CMS2 in β-C3P was found, with a solubility limit of ∼19%.
García-Carrodeguas et al. [7] revisited this system, which they considered as pseudo-binary, confirming the presence in it of a eutectic point located at 63% CMS2–37% C3P, at a temperature of 1300±5°C. These researchers found that at the eutectic point the CMS2 phase actually coexists with a solid solution of CMS2 in C3P, which they denominated as β-C3Pss. In this solid solution, there is a partial substitution of Mg for Ca, as well as a partial substitution of Si for P, and it can be represented either by β-[(Ca1−xMgx)3(P1−δSiδO4−δ/2)2] or β-[Ca3−xMgx(P1−δSiδO4)2□δ] chemical formula [7,8], where □ denotes oxygen vacancies created by the charge compensation required for the substitution of Si4+ for P5+. Since the dissolution of CMS2 in C3P is incongruent, the formation of the β-C3Pss solid solution produces a small amount of segregated SiO2 in the material, probably in the form of amorphous nano-clusters. Thus, the system is actually ternary, although for practical purposes it may be considered to be binary due to the small amount of segregated SiO2 present in it [7]. These researchers did not detect the formation of solid solutions of C3P in CMS2, but they suggested that part of the segregated SiO2 could be incorporated into the crystal lattice of CMS2. In this system, the microstructure of the material with eutectic composition is composed by alternate layers of CMS2 and β-C3Pss [7].
In the present study, a glass-ceramic material of slightly hypereutectic composition of the pseudo-binary system CMS2–C3P was synthesized by the petrurgic route, in which the primary crystallization of a melt occurs during slow cooling of the latter from its liquidus temperature to room temperature, using a single melting and cooling cycle [9]. It was aimed to achieve an in situ formation of the CMS2 and β-C3Pss phases in a single step, as well as to obtain in the synthesized materials a microstructure consisting of β-C3Pss primary phase embedded in a matrix of the CMS2-β-C3Pss eutectic phase. A further purpose of the work was to verify that the in vitro bioactivity behavior of the latter materials was similar to that shown by Bioeutectic® [5], since their microstructure was significantly different to that of the material of eutectic composition of the same system synthesized by the solid-state reaction process [7,10,11]. The results obtained from the synthesis of the studied materials are presented in this work, while the results obtained from the in vitro bioactivity tests carried out for the same materials will be presented separately.
Experimental procedureThe studied material was synthesized using a procedure based on that reported for the synthesis of Bioeutectic® [5]. We studied the composition considered by Sata [6] as the eutectic composition of system CMS2–C3P (61% CMS2–39% C3P), but which is currently known [7] to be slightly hypereutectic, falling within the primary crystallization region of the β-C3Pss solid solution.
An initial mixture of 41.13% CaCO3, 20.69% (NH4)2HPO4, 21.12% SiO2, and 17% (MgCO3)4·Mg(OH)2·5H2O, all of them reagent-grade products from Aldrich and Spectrum brands, was prepared and homogenized. All materials were used as received, without further purification. After elimination of the gaseous species (CO2, NH3 and H2O) generated by decomposition at high temperature of some of the precursor reagents, the mixture had the following oxide composition: 36.94% CaO, 17.85% P2O5, 33.85% SiO2 and 11.35% MgO. The average particle size of the reagents was in the range of ∼8–10μm. The reactions proposed for the in situ synthesis of the desired phases are:
These reactions were used to calculate the relative proportion in the initial mixture of each one of the precursor reagents in order to obtain a material with the desired composition in the system CMS2–C3P. The mixture was melted at a temperature of 1450°C in a 75-ml platinum crucible, using a heating rate of 5°C/min in a Lindberg/blue M electric furnace, model BF51433 PC-1. The crucible was placed at the center of the furnace chamber resting on the floor of the furnace, near the furnace thermocouple. After holding it for 2h at the mentioned temperature, the melt was cooled down to 1320°C at a rate of 3°C/min, which is a temperature slightly higher than the liquidus temperature of the studied composition [7]. Upon reaching this temperature, the cooling rate was reduced to 0.5, 1 or 2°C/h, in order to slowly cool the molten material through the mushy zone until the eutectic temperature of the system (1300°C) was reached. Upon reaching this temperature, the cooling rate was changed back to 5°C/min and the cooling was continued until room temperature was attained. The cooling stage in the temperature range of 1320–1300°C lasted for 40, 20 and 10h, for the cooling rates of 0.5, 1 and 2°C/h through the mushy zone, respectively. The crystallized material was extracted from the crucible by fracturing it mechanically or by thermal shock treatment (sudden heating at 700°C, followed by quenching of the crucible bottom in cold water). This extraction procedure did not modify the microstructure of the crystallized materials.
The synthesized materials were characterized using X-ray diffraction (XRD) and scanning electron microscopy (SEM) techniques. For the XRD analyses, monochromatic Cu Kα radiation was used in a Philips X’Pert 3040 device, with a scanning speed of 0.03°/s in the 2θ range from 20° to 50°. Prior to the analyses carried out on the SEM, the samples were mounted using a fast-setting epoxy resin, and then ground and polished using standard ceramographic techniques, giving them a final mirror polish with diamond paste with successive particle sizes of 3, 1 and 0.5μm. Finally, the samples were cleaned, dried and coated with a graphite film using a JEOL JEE-400 vacuum evaporator. For the observation on the SEM, a Philips XL30 ESEM device with an acceleration voltage of 20kV and a working distance of 10mm was used. The specimens used for the characterization analyses were taken from the central zone of the crucible, away from its walls.
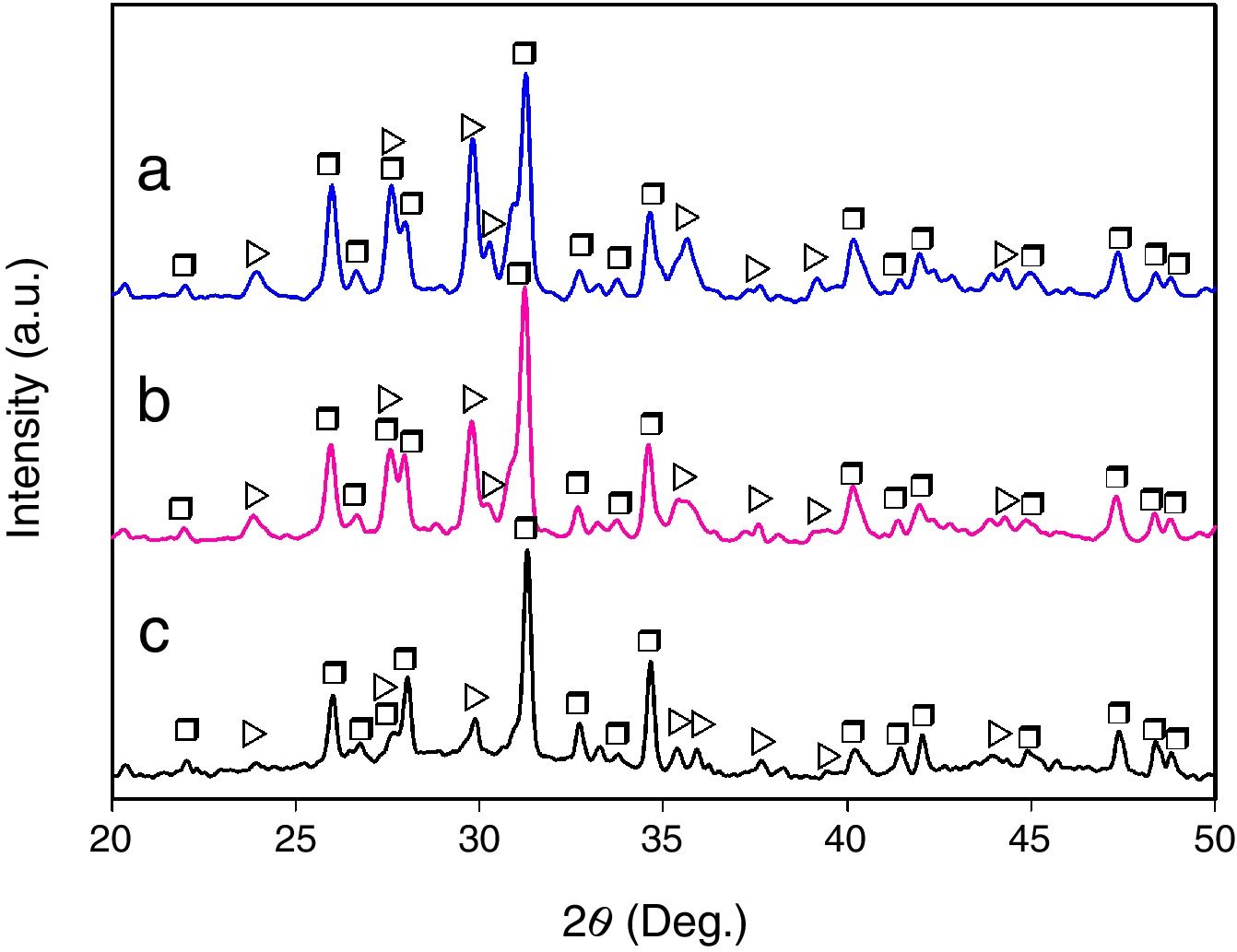
Results and discussionMicrostructural evolutionFig. 1 shows the XRD patterns corresponding to the glass-ceramic materials synthesized using cooling rates of 0.5, 1 and 2°C/h through the mushy zone. As it can be seen, in all cases only the presence of the CMS2 and β-C3Pss phases was detected, observing in a qualitative way, based on the behavior of the relative intensity of the peaks, an increase in the relative proportion of the first phase, as well as a decrease in the relative proportion of the second one, with increasing cooling rate. Probably the presence of the α-C3Pss phase was not detected due to the α-C3Pss→β-C3Pss transformation taking place during cooling of the samples [7]. On the other hand, it is well demonstrated [12] that small amounts of Si in C3P stabilize its high temperature form (α-C3P), which then can be found at room temperature as a metastable phase. The fact that this phase is not stabilized in the system CMS2–C3P, could be related to the substitution of Mg for Ca and Si for P that takes place simultaneously during formation of the β-C3Pss phase.
 2, (b) 1, and (c) 0.5°C/h. Key: ▷ CMS2 (JCPDS card no. 98-003-0522), and □ β-C3Pss (JCPDS card no. 01-070-0682).")
The presence of an amorphous halo was clearly detected in the XRD pattern obtained for the sample cooled at 0.5°C/h (Fig. 1c), which was located approximately in the 2θ range between 25° and 35°, and which was attributed to the presence in the material of a glassy phase segregated during formation of the β-C3Pss phase. In the case of the other two cooling rates used, the intensity of the amorphous halo decreased markedly in the corresponding XRD patterns with increasing cooling rate (Fig. 1a and b), which was likely due to a decrease in the proportion of the segregated glassy phase in the samples.
Figs. 2 and 3 show the SEM micrographs obtained for the same samples. The microstructures of all materials were very similar to each other, with the presence of long dendrites of the β-C3Pss primary phase, whose range of length and thickness increased with decreasing cooling rate; the longest dendrites obtained for the cooling rate of 0.5°C/h had a length of ∼3mm (see Table 1). The primary dendrites were embedded in a matrix constituted by a eutectic phase formed by alternate lamellar layers of CMS2 and β-C3Pss phases. There was also a likely additional presence in the matrix of segregated CaO–SiO2 nanoaggregates (see section “Effect of cooling rate on phase composition of the synthesized glass-ceramics”), which were probably not observed on the SEM due to their fine size.
![SEM micrographs of samples cooled through the mushy zone at rates of (a) 2, (b) 1, (c) and (d) 0.5°C/h [micrograph (d) was taken at lower magnification]. Primary dendrites of the β-C3Pss phase (light gray) can be seen embedded in a eutectic matrix (dark gray).](https://static.elsevier.es/multimedia/03663175/0000006000000002/v1_202104020823/S0366317520300169/v1_202104020823/en/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNd3TCEZuNxhudow/+3Hrh9WJc+mtnYSSPL9K6mfMWVj1q8jQe17dJd7SXrwa74/LlNDNWQ627ya46iuDvk+eEoTl5E5uaFQayuSHP1uaSpPlIOJ5wrQTwR6h2zPeRjA9aTQn0ZTP6MCYI52N+8NFwV2eBwVv+eERUj3942L2KxgeSy6YkEKAY1Q/1e3orQogh3vEW3qCIgNAHbtX29KFaIdZX7Ta19vdtIdPz92QgLceWK0QKgbmPsEtTCfcRTa/vo= "SEM micrographs of samples cooled through the mushy zone at rates of (a) 2, (b) 1, (c) and (d) 0.5°C/h [micrograph (d) was taken at lower magnification]. Primary dendrites of the β-C3Pss phase (light gray) can be seen embedded in a eutectic matrix (dark gray).")
 and Ca, P, Mg and Si X-ray distribution maps obtained for a sample cooled at a rate of 0.5°C/h through the mushy zone.")
The morphology of the interdendritic eutectic phase observed in this work is similar to that reported by García-Carrodeguas et al. [7] for a material of eutectic composition of the pseudo-binary system CMS2–β-C3P. These researchers observed a microstructure composed by alternate layers of CMS2 and β-C3Pss phases for the latter material melted at 1400°C/2h and then cooled down to 1300°C (eutectic point of the system), at a cooling rate of 1°C/h. Although the β-C3Pss layers seem to be discontinuous in the eutectic phase (see Fig. 4b of Ref. [7]), however, they are in fact continuous, with an actual morphology corresponding to ribbons or plates, as pointed out by Elliott [13].
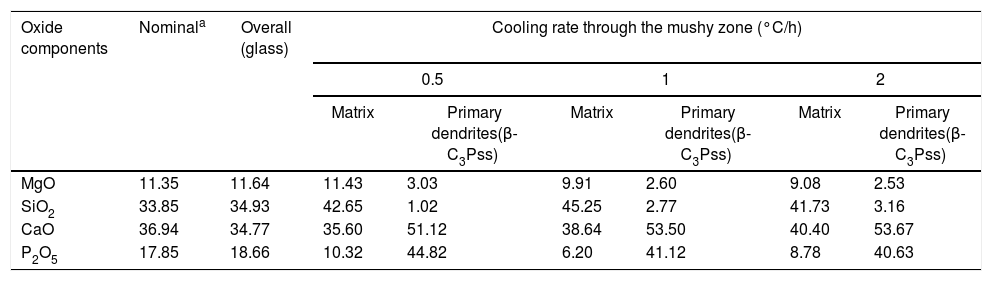
The identification of the phases constituting the matrix and the long dendrites was made based on the EDS spectra (Table 2) and the X-ray maps (Fig. 3) obtained on the SEM for the sample synthesized using a cooling rate of 0.5°C/h. In all cases, observations at low magnification (Fig. 2d) allowed us to visualize a large number of dendrite colonies of the β-C3Pss primary phase, each one of which originated at a point near the wall of the platinum crucible, where nucleation took place, and which grew radially toward the center of the crucible. This suggested that platinum acted as a nucleating agent during the melt crystallization [14]. While a small tilting angle existed between neighboring dendrite colonies, within each colony the dendrites grew almost parallel to each other.
Compositions (%) determined by EDS analyses on the SEM for the synthesized glass-ceramic materials.
| Oxide components | Nominala | Overall (glass) | Cooling rate through the mushy zone (°C/h) | |||||
|---|---|---|---|---|---|---|---|---|
| 0.5 | 1 | 2 | ||||||
| Matrix | Primary dendrites(β-C3Pss) | Matrix | Primary dendrites(β-C3Pss) | Matrix | Primary dendrites(β-C3Pss) | |||
| MgO | 11.35 | 11.64 | 11.43 | 3.03 | 9.91 | 2.60 | 9.08 | 2.53 |
| SiO2 | 33.85 | 34.93 | 42.65 | 1.02 | 45.25 | 2.77 | 41.73 | 3.16 |
| CaO | 36.94 | 34.77 | 35.60 | 51.12 | 38.64 | 53.50 | 40.40 | 53.67 |
| P2O5 | 17.85 | 18.66 | 10.32 | 44.82 | 6.20 | 41.12 | 8.78 | 40.63 |
The microstructure of the material was the result of the way it was cooled inside the furnace, and it can be explained based on the following considerations. During the solidification of a non-eutectic binary composition a destabilization of the solid/liquid planar (lamellar) interface may take place during the growth of the eutectic phase, resulting in the formation of a microstructure composed by primary dendrites plus interdendritic binary eutectic phase [15,16]. The morphological instability of the planar solidification front is caused by the rejection of solute ahead of it, which modifies the temperature of the solid/liquid interface. This causes the primary phase to become strongly constitutionally undercooled, which makes it to grow faster, or at a higher temperature, than the eutectic phase.
Under the cooling conditions that prevailed inside the furnace, crystallization began on the inside of the Pt crucible walls, probably aided by a nucleating effect of said metal, as mentioned before, and the primary crystals grew in the form of elongated dendrites. According to the literature [17], the direction of growth of the primary or main dendrite arms depends on two main factors. On the one hand, dendrites grow along one of the preferred crystallographic axes of growth, which depends on the lattice structure of the material. On the other hand, the direction of growth of dendrites is parallel to the direction of heat flow, but in the opposite direction to it. Therefore, the primary arms of the dendrites grew from the inner surface of the crucible wall, in the direction of the center of the molten material, also in a radial manner but in a direction opposite to that of the heat extraction.
It was observed that the dendrites became smaller and thicker as the cooling rate decreased (Fig. 2). This is related to the way in which the dendrites nucleate and grow as a function of cooling rate [15–17]. In an undisturbed liquid, a slow cooling rate results in the nucleation of a small number of dendrites, and under these conditions, the dendrites can become considerably coarser. This could explain the increment in the relative proportion of the β-C3Pss phase that was observed in the XRD patterns with decreasing cooling rate (Fig. 1). Since it is also known that the interdendritic spaces increase with increasing cooling rate, at 1 and 2°C/h a greater separation between the dendrites was observed, with respect to what was seen in the sample crystallized at 0.5°C/h.
Independently of the cooling rate used, the primary dendrite arms of the β-C3Pss phase showed a characteristic “skeletal” shape, see Figs. 2 and 3. This morphology is typical of crystals formed under non-equilibrium conditions with diffusion-controlled fast growth rates, which is promoted by a high degree of supersaturation, as well as by a high degree of undercooling, in the melt [18]. The undercooling degree becomes more pronounced with increasing cooling rate [19]. Skeletal crystals are relatively common in igneous rocks such as olivine [20]. They are characterized by a series of discontinuities, or entries, which are filled with the matrix material, and which are formed due to a slower growth rate, and a lower supersaturation, existing in the areas where such discontinuities appear, with respect to the rest of the crystal [21,22]. This is frequently called the “Berg effect” [23,24]. The growth rate of the skeletal crystals is relatively rapid, even at slow cooling rates, as long as the liquid is substantially undercooled before nucleation [25]. A delayed nucleation contributes to the strong supersaturation necessary for the rapid growth of the skeletal crystals [26]. No examples were found reported in the literature regarding the β-C3Pss phase crystals showing a skeletal morphology. However, since it is known that HAp containing low to moderate levels of Si can adopt this type of crystal morphology [27], thus, it is perhaps not surprising that β-C3Pss could also be able to do so under the appropriate conditions. A microstructure consisting of skeletal primary crystals surrounded by a lamellar eutectic phase has also been observed for the case of the eutectic composition of the system Ni31Si12–Ni2Si, when the corresponding melt was solidified under undisturbed conditions (see Fig. 2a of Ref. [28]).
Lastly, the dendrites of the β-C3Pss primary phase showed a very peculiar morphology, which was characterized by growth of their secondary arms in a preferred downwards direction. This conferred a comb-like shape to them due to the suppression of the growth of the secondary dendritic arms in any other direction. This could be attributed to the effect of competitive growth between neighboring dendrites [29–31], as well as to a diffusion-limited supersaturated environment [32]. The comb-like morphology of the dendrites was most clearly observed after soaking the samples for 7 days in a Simulated Body Fluid (SBF, saline solution that simulates the chemical composition of human blood plasma), since after this soaking time the material's matrix was dissolved into the SBF, exposing the nude dendrites, see Fig. 4. Perhaps the preferential downward growth of the secondary dendritic arms was the result of a lower rate of cooling of the material through the bottom of the crucible.
Effect of cooling rate on phase composition of the synthesized glass-ceramics
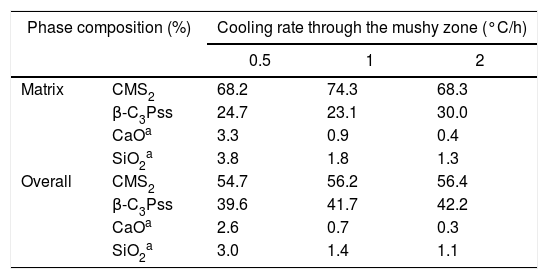
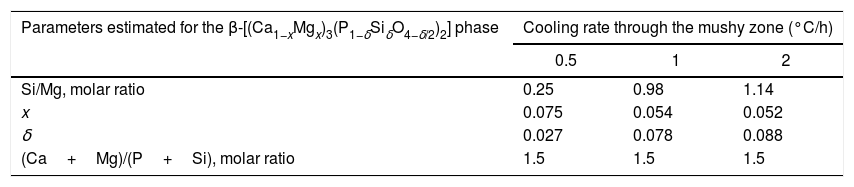
From the nominal composition of the synthesized materials, as well as from the results of the EDS analyses performed (Table 2), we estimated the overall and the matrix phase compositions (Table 3), as well as the level of substitution of Mg for Ca (x) and that of Si for P (δ), and the Si/Mg and (Ca+Mg)/(P+Si) molar ratios in the β-C3Pss primary phase (Table 4). It was assumed that the latter phase has the same composition in both the interdendritic region and the primary dendrites. It was also assumed that all the P2O5 present in the matrix is contained in the β-C3Pss phase existing in the same region, and that the dissolution of CMS2 into C3P is incongruent, in such a way that the Si/Mg molar ratio of the β-C3Pss phase differs from that of the CMS2 phase [7]. The possibility that CMS2 forms solid solutions with silica [7] was completely ruled out, since no evidence of the occurrence of this phenomenon was found. The calculations were made assuming that the formation of the β-C3Pss solid solution from the CMS2 and C3P phases obeys reaction (3).
Compositions estimated from the results obtained by EDS on the SEM for the crystallized materials.
Composition (molar %) and other parameters of the β-C3Pss phase estimated from the results of the EDS analyses carried out on the SEM.
| Parameters estimated for the β-[(Ca1−xMgx)3(P1−δSiδO4−δ/2)2] phase | Cooling rate through the mushy zone (°C/h) | ||
|---|---|---|---|
| 0.5 | 1 | 2 | |
| Si/Mg, molar ratio | 0.25 | 0.98 | 1.14 |
| x | 0.075 | 0.054 | 0.052 |
| δ | 0.027 | 0.078 | 0.088 |
| (Ca+Mg)/(P+Si), molar ratio | 1.5 | 1.5 | 1.5 |
It is worth mentioning that García-Carrodeguas et al. [7] claimed that the incongruent dissolution of CMS2 into C3P causes the segregation of amorphous nano-clusters of SiO2 in the material. However, our analyses indicate that it is most likely that these nano-clusters are constituted by a glass of the system CaO–SiO2 (reaction (3)), and that the statement of these researchers corresponds to the particular case in which 2x=δ. However, in general, we consider that this situation is very unlikely, and thus, a co-segregation of CaO and SiO2 should take place during the formation of the β-C3Pss phase.
In the results presented in Table 4 it can be seen that, in all cases, the composition of the matrix presents a CMS2/β-C3Pss weight ratio that differs significantly from the CMS2/β-C3P=63/37 value reported by García-Carrodeguas et al. [7] for the eutectic phase of the pseudo-binary system CMS2–C3P. In general, it would be expected that CMS2/β-C3Pss≅CMS2/β-C3P for the eutectic phase, due to the relatively small value of the x and δ substitution levels. The CMS2/β-C3Pss weight ratio of our materials varied depending on the cooling rate. Sharp and Flemings [33] mentioned that in the unidirectional solidification regimen the composition of the interdendritic “eutectic” varies according to the G/R ratio (thermal gradient/growth rate), which is why it differs from that of the eutectic phase of the corresponding phase diagram. A similar phenomenon could have occurred in the present work, due to the way in which the primary dendrites grew (see section “Microstructural evolution”).
On the other hand, Tables 3 and 4 indicate that among the different parameters estimated from the results obtained by EDS, the increment in the cooling rate had the greatest effect on the x and δ substitution levels and, consequently, on the Si/Mg molar ratio of the β-C3Pss phase. It was observed that the value of x decreased from 0.075 to 0.052 when the cooling rate was increased from 0.5 to 2°C/h. In contrast, the value of δ increased from 0.027 to 0.088 with the same increment in the cooling rate. In other words, the greatest Mg for Ca substitution level corresponded to the lowest cooling rate, while the greatest Si for P substitution level corresponded to the fastest cooling rate. This reflects the fact that the concentration of MgO in the β-C3Pss phase decreased from a value of 3.03% at 0.5°C/h to a value of 2.53% at 2.0°C/h, while the concentration of SiO2 in this phase was increased from 1.02% to 3.16% for the same increment in the cooling rate (Table 2). Consequently, the Si/Mg molar ratio was increased in the β-C3Pss phase from a value of 0.25 at 0.5°C/h to a value of 1.14 at 2°C/h (Table 4). The fact that in all cases the Si/Mg molar ratio of the β-C3Pss phase was smaller than two, evidenced the occurrence of incongruent dissolution of CMS2 into the C3P crystal lattice [7]. On the other hand, as already mentioned, the substitution of Si4+ for P5+ requires the creation of oxygen vacancies in the solid solution in order to compensate the charges. It has been reported [34] that the concentration of oxygen vacancies can be increased by increasing the cooling rate. Therefore, the cooling rates of 1 and 2°C/h could have promoted a greater creation of oxygen vacancies at the moment of the substitution of Si4+ for P5+, with respect to the cooling rate of 0.5°C/h, and this provoked that the mentioned substitution was easier when the two faster cooling rates were employed. This is supported by the fact that it was possible to reach a concentration of ∼1.4molar % Si in the β-C3Pss phase of the sample with the fastest cooling rate (2°C/h), which is within the range of 1.2–3.9molar % Si mentioned by García-Carrodeguas et al. [7] for the saturation of β-C3Pss with silicon. This also agrees with the observations made regarding the segregation of SiO2 into the material's matrix during the formation of the β-C3Pss phase (Table 3). A higher incorporation of silicon in this phase implied a lower segregation of SiO2 into the material's matrix. The lowest segregation of SiO2 into the matrix was obtained for the cooling rate of 2°C/h, which also resulted in the highest value of δ in the β-C3Pss phase. When the cooling rate was increased from 0.5 to 2°C/h, there was a trend toward a decrement in the amount of segregated CaO and SiO2, which was from 2.6% to 0.3% in the first case and from 3.0% to 1.1% in the second case. All these results are consistent with those obtained by XRD (see Fig. 1 and section “Microstructural evolution”). Finally, as expected according to the chemical formula of the β-C3Pss phase, its (Ca+Mg)/(P+Si) molar ratio had a constant value equal to 1.5, independently of the x and δ substitution levels and cooling rate (Table 4).
It is worth mentioning that it is known [35] that SiO2 increases the bioactivity of calcium phosphates, therefore, the presence of a small amount of this oxide segregated in the matrix of the studied materials, in the form of a glassy phase of the system CaO–SiO2, may have a positive effect on their bioactivity.
One final remark. In order to fully appreciate the figures in color included in this paper, we encourage the reader to see them in the digital version of the work, since they may appear in black and white in its printed version.
ConclusionsIn general, the microstructure obtained in the synthesized materials with composition 61% CMS2–39% C3P consisted of β-C3Pss primary dendrites embedded in a matrix of lamellar CMS2–β-C3Pss eutectic phase, which also presumably contained a small amount of segregated CaO–SiO2 glass. The microstructure and phase composition of the synthesized glass-ceramic materials were affected by the cooling rate used through the mushy zone during crystallization of the samples. Dendrite colonies of the β-C3Pss primary phase originated on the inner wall of the platinum crucible, growing radially toward the center of it. A small tilting angle existed between neighboring dendrite colonies, but the dendrites grew almost parallel to each other within each colony. The formation of this microstructure was attributed to the strong constitutional undercooling achieved by the primary phase, which allowed it to grow faster than the eutectic phase. The dendrites became smaller and coarser, and the interdendritic space decreased, as the cooling rate decreased. This was related to the way in which the dendrites nucleate and grow as a function of cooling rate. The comb-like morphology shown by the dendrites of the primary phase, with their secondary arms growing in a preferred downwards direction, was attributed to a competitive growth between neighboring dendrites as well as to a diffusion-limited supersaturated environment. Independently of the cooling rate, the crystals of the primary phase showed a “skeletal” morphology characterized by a series of discontinuities, or entries, filled with the matrix material, and which were formed under non-equilibrium conditions and diffusion-controlled fast growth rates. Due to the way in which the dendrites grew, it was hypothesized that the composition of the interdendritic eutectic phase varied according to the G/R ratio (thermal gradient/growth rate), and thus, it significantly differed from that of the eutectic phase of the corresponding phase diagram. Lastly, it was found that the greatest Mg for Ca substitution level was attained for the primary phase formed in the samples crystallized using the lowest cooling rate, while the greatest Si for P substitution level was achieved in the primary phase formed in the samples crystallized using the fastest cooling rate. This was attributed to an easier creation of oxygen vacancies during the substitution of Si4+ for P5+ in the primary phase, in the latter samples.
Authors’ contributionJ. López-Cuevas ensures that all authors are included in the author list, its order has been agreed by all authors, and that all authors are aware that the paper was submitted. He originated the main idea of the research work and supervised the conducted experimental work, participating in every aspect of it. He also wrote the draft paper and prepared the final manuscript.
C.M. López-Badillo carried out the experimental work, and contributed to the analysis and discussion of the results and writing of the paper.
J. Méndez-Nonell co-directed the experimental work, assisted in every aspect of the experimental and characterization work, and supervised the final manuscript.
Conflict of interestThe authors declare that they have no conflict of interest.
J.L.C. thanks SEP and Cinvestav for the funding granted for the realization of this work (Cinvestav's Scientific Research and Technological Development Fund, Project No. 252). C.M.L.B. also thanks CONACYT for the scholarship granted for the completion of her Master of Science studies at Cinvestav-Saltillo, México. This paper is based on part of her M.Sc. thesis work.

