





Inflammatory pseudotumor (IPT) and inflammatory myofibroblastic tumor (IMT) are two very rare entities that were formerly included in the same category; however, today they are considered two different diseases due to the neoplastic origin of the IMT. Our objective is to share our experience in the management of these two types of tumors that we must take into account in the differential diagnosis of pulmonary masses or nodules.
MethodsThirteen patients with a pathological diagnosis of IPT and IMT who underwent surgery between 2008 and 2019 were retrospectively studied. We recorded the pre and postoperative information of each one, as well as the survival analysis.
ResultsOf the 13 patients, 8 were men and 5 women. The mean age of presentation was 53,5 years. An atypical segmentectomy was performed in 6 patients; a lobectomy was necessary in 6 and a pneumonectomy in 1 case. In all cases a complete resection was achieved. Diagnosis was possible thanks to histology, immunohistochemical (IHQ) and fluorescent in situ hybridization (FISH) techniques determining the expression of IgG4 and the rearrangement of ALK, respectively. After a median follow up of 49 months, we didn’t find any loco-regional or distant recurrence in the patients studied.
ConclusionIPT and IMT are rare tumors with a very good prognostic. The diagnosis of both entities is based mainly on specific anatomopathological techniques. Surgery has, in most cases, both a diagnostic and therapeutic role.
El pseudotumor inflamatorio (PTI) y el tumor miofibroblástico inflamatorio (TMI) son dos entidades muy poco frecuentes que se incluían antiguamente en la misma categoría; sin embargo, en la actualidad se consideran dos enfermedades diferentes debido al origen neoplásico del TMI. Nuestro objetivo es compartir nuestra experiencia en el manejo de estos dos tipos de lesiones que debemos tener en cuenta en el diagnóstico diferencial de masas o nódulos pulmonares.
MétodosFueron estudiados retrospectivamente 13 pacientes con diagnóstico anatomopatológico de PTI o TMI intervenidos entre los años 2008 y 2019. Registramos la información pre y postoperatoria de cada uno, así como el análisis de supervivencia.
ResultadosDe los 13 pacientes, 8 eran varones y 5 mujeres. La media de la edad de presentación fue de 53,5 años. En 6 pacientes se practicó una segmentectomía atípica, en 6 fue necesario realizar una lobectomía y en 1 caso una neumonectomía. En todos los casos se consiguió una resección completa. El diagnóstico fue posible gracias a la histología, técnicas de inmunohistoquímica (IHQ) y de hibridación fluorescente in situ (FISH) determinando la expresión de IgG4 y el reordenamiento de ALK, respectivamente. Tras una mediana de seguimiento de 49 meses no se observaron datos de recidiva locorregional ni a distancia en los pacientes estudiados.
ConclusiónEl PTI y el TMI son tumores poco frecuentes con muy buen pronóstico. El diagnóstico de ambas entidades se basa principalmente en técnicas anatomopatológicas específicas. La cirugía tiene, en la mayor parte de las ocasiones, un papel tanto diagnóstico como terapéutico.

The term ‘inflammatory pseudotumor’ (IPT) was first described in 19391 to identify a heterogeneous group of fibroinflammatory diseases, both reactive and neoplastic in origin. Since then, these tumors have received many names (plasma cell granuloma, pseudosarcomatous fibromyxoid tumor, inflammatory myofibrohistiocytic proliferation, etc)2,3, all of which reflect their varied cellular composition. Different hypotheses have been posed about the origin of these lesions. Melloni et al.4 suggest a disproportionate inflammatory reaction to an underlying infectious process. Thus, the possible association with the Epstein-Barr virus5,6 and herpes virus type 87 has also been contemplated.
Inflammatory myofibroblastic tumors (IMT) are characterized by a proliferation of myofibroblasts that are closely related to an inflammatory infiltrate of plasma cells, lymphocytes and eosinophils8. They may occur in any anatomical region but show a predilection for the lungs, soft tissue, and abdominopelvic cavity of children and young adults9,10. In the past, IMT were classified as benign lesions, although they are currently considered neoplasms of intermediate biological potential due to their ability to recur and produce metastasis3.
In the past, both IPT and IMT were included in the same category due to similar morphological characteristics from a pathological standpoint, such as fibroblastic-myofibroblastic proliferation and the presence of inflammatory cells. In fact, recently published studies have frequently confused the 2 entities11,12. Currently, however, they are considered 2 different tumors due to the neoplastic origin of IMT3. Given the small number of cases published in the literature to date, we present our experience from the last 11 years based on a series of patients diagnosed with IPT and IMT who were treated surgically by our service, analyzing diagnosis, treatment and survival.
MethodsFrom 2008 to 2019, we operated on a total of 13 patients with the final diagnosis of either IPT or IMT. All patients underwent a preoperative fiberoptic bronchoscopy examination. In cases of peripheral location, transthoracic fine-needle aspiration (FNA) was performed for diagnosis. In the 13 cases, the definitive diagnosis of IPT or IMT was reached by pathological study of the resected piece. The data used in the study were collected retrospectively in a coded and anonymized computer database, complying with current Spanish legislation regarding personal data protection and after having received the approval of the hospital ethics committee.
The following variables were studied: age, sex, comorbidity (tobacco consumption and cancer history), clinical manifestations, location of the lesion and morphology on computed tomography (CT) scan, size of the lesion in millimeters, standardized uptake value (SUV) on positron emission tomography (PET), performance of FNA or bronchoscopy, surgical approach (video-assisted surgery or thoracotomy/sternotomy), type of surgical resection (lobectomy, atypical resection or pneumonectomy), postoperative hospital stay, postoperative complications, immunohistochemical analysis (vimentin, desmin, cytokeratin, smooth muscle actin, EMA, IgG4, and ALK), molecular biology techniques (FISH), and survival.
The statistical analysis was performed using the SPSS® v.24.0 for Windows. Global results are expressed as absolute and relative frequencies for qualitative variables. For quantitative variables, the mean, median and range were used. A P-value <.05 was considered statistically significant.
ResultsAmong the 13 patients included, 9 cases were pathologically diagnosed as IPT and 4 as IMT. There were 8 males (61.53%) and 5 females (38.46%), with a predominance of males in the IPT group (6/3). The mean age of presentation in the series was 53.5 years, with a median of 48 years (range: 11–82 years), which was 60.1 years in the IPT group and 47.8 years in the IMT group (P = .243). The analysis of comorbidities identified 6 patients (46.15%) who were smokers at the time of diagnosis, while 2 patients had a history of cancer (germ cell tumor and pancreatic gastrinoma).
With regards to clinical presentation, only 2 patients were symptomatic at the time of diagnosis (one case of IMT with dyspnea and one case of IPT with hemoptysis), while in the remaining cases the finding was incidental. No significant laboratory abnormalities were detected in any of the patients studied. By analyzing the radiological characteristics of these tumors on CT, we were able to verify that the lesions were predominantly solid with smooth edges (12/13 cases), and only one case presented cavitation; the median tumor size measured radiologically was 22 mm (range: 8–135 mm). No patient presented signs of tumor invasion of neighboring structures on the imaging studies. PET showed pathological uptake in all the patients studied, with a mean maximum standardized uptake value (SUVmax) of 4.3 g/mL for IPT and 2.6 for IMT (P = .198). The lesions were mainly located in the lower lobes (4 left and 3 right), with a predominance of IPT (71.42%). Although endoscopic samples were taken in 12 of the 13 patients who underwent fiberoptic bronchoscopy (92.30%), the sample was insufficient to reach a diagnosis in all cases. FNA was performed in patients with peripherally located lesions, although it was not diagnostic in any of the cases.
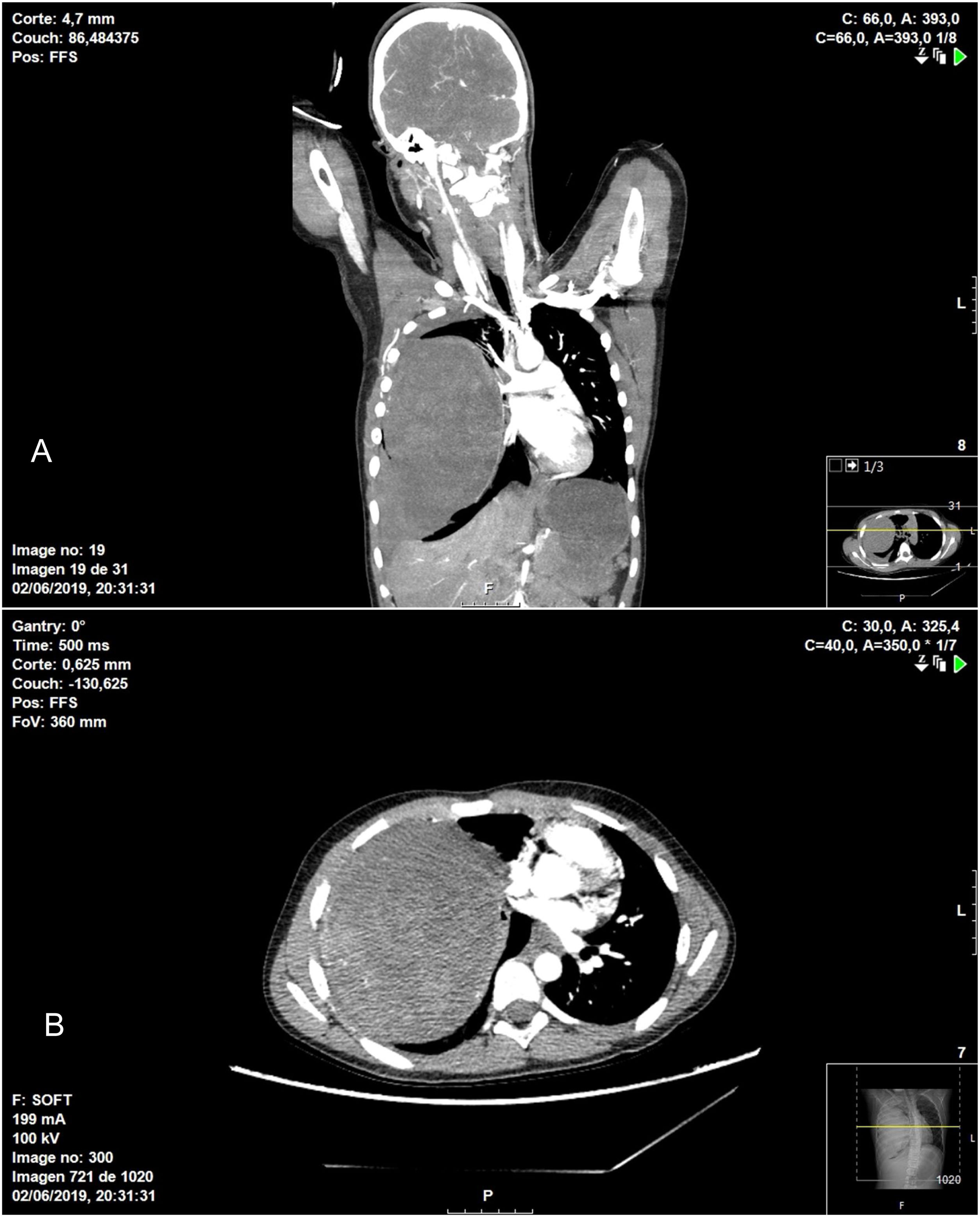
In terms of the surgical technique performed, 6 patients underwent wedge resection (4 IPT and 2 IMT) after intraoperative pathological confirmation of the mass, which was reported as benign disease. In 6 patients, lobectomy was necessary due to the location of the lesion (4 IPT and 2 IMT), and in one patient (IPT) right pneumonectomy was performed through a median sternotomy. This case was an 11-year-old patient who was treated surgically in collaboration with the pediatric surgery service due to his clinical presentation (hemoptysis and hemorrhagic shock), as the thoracic CT scan had revealed a large tumor occupying the entire right hemithorax with signs of intratumoral bleeding, hemothorax and mediastinal displacement (Fig. 1).
 coronal image; B) axial image.")
Chest CT showing heterogenous intrapulmonary mass associated with moderate hemothorax occupying practically the entire right hemithorax, causing significant mass effect, with complete collapse of the ipsilateral pulmonary vessels in addition to displaced mediastinal structures: A) coronal image; B) axial image.
Complete tumor resection was achieved in all cases. The most used approach was video-assisted surgery (61.53%). Mean postoperative hospital stay was 3.2 days (range: 3–6 days), with no postoperative complications during this period.
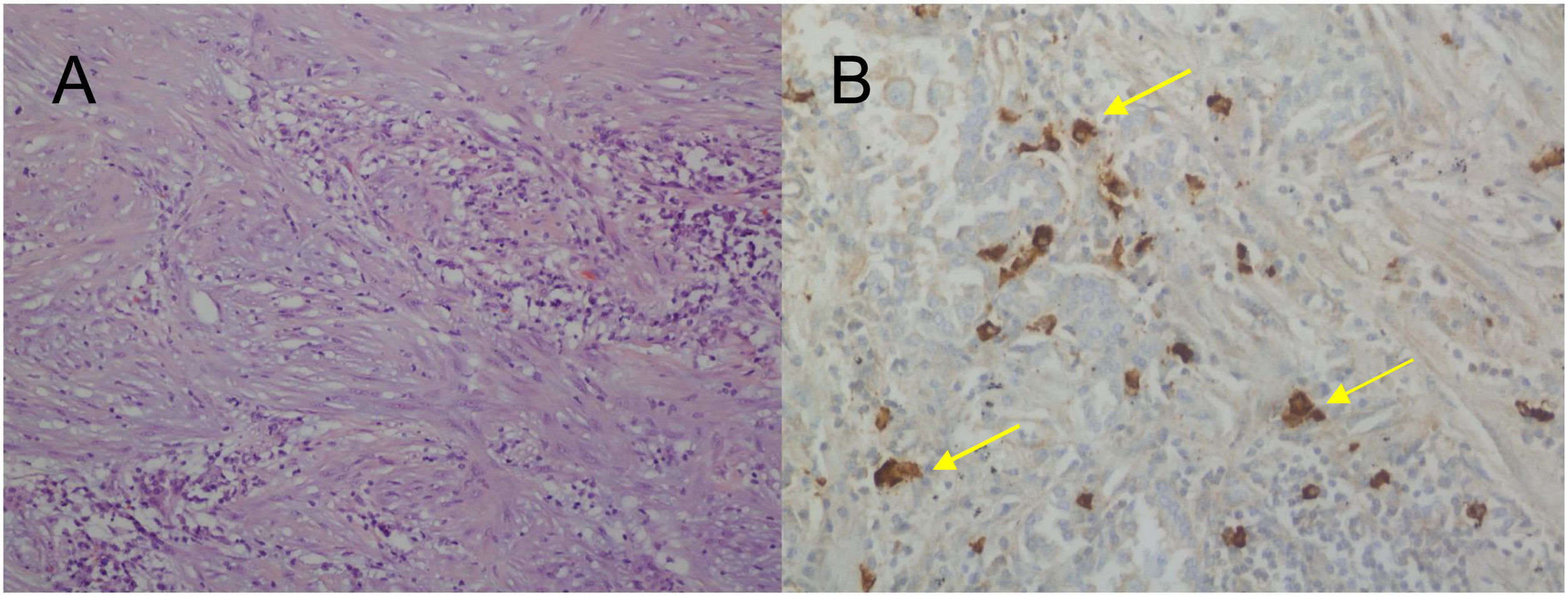
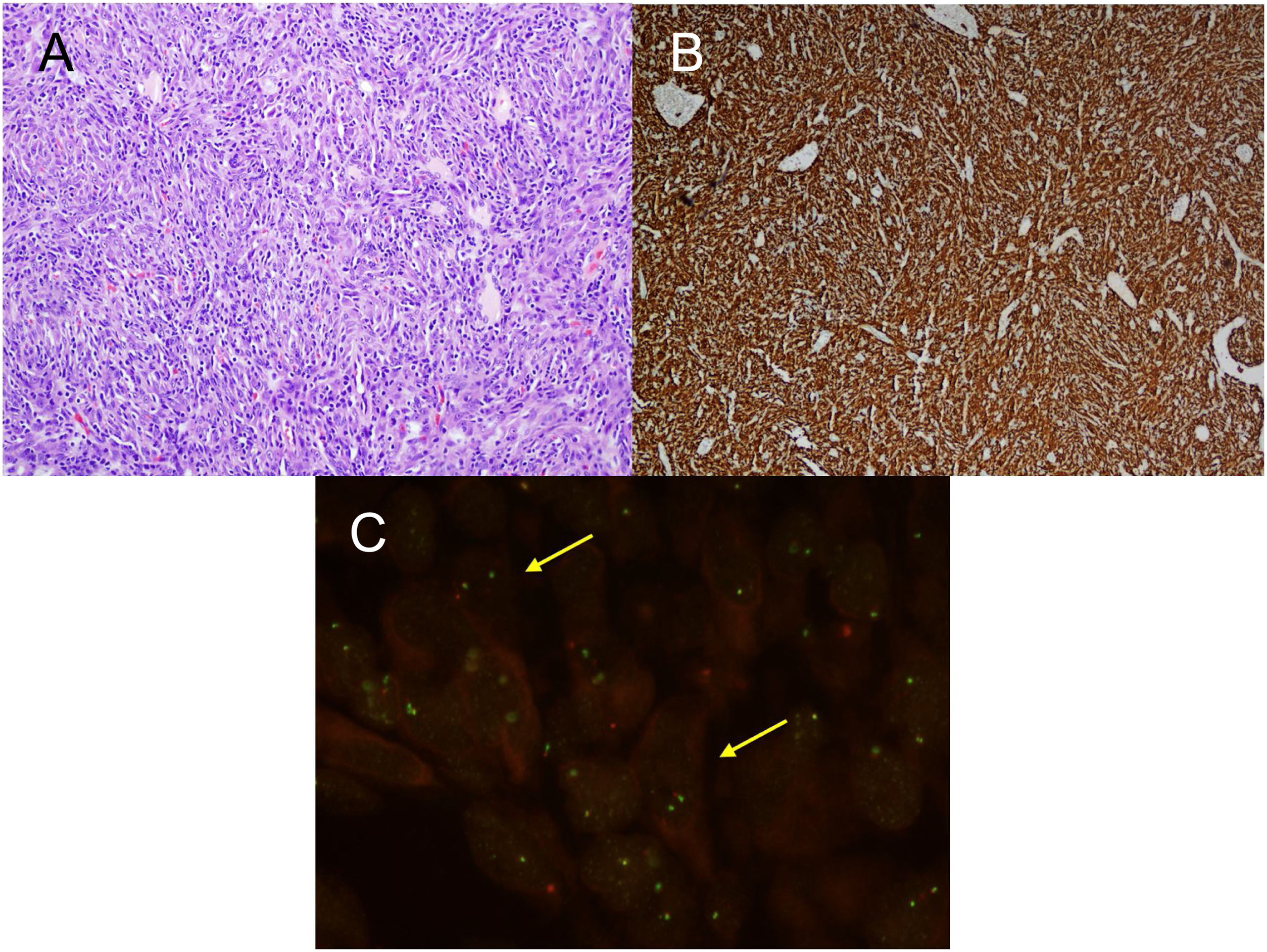
For the pathological study of the resection pieces, tissue sections fixed in formaldehyde and embedded in paraffin were used. Hematoxylin-eosin staining and immunohistochemical (IHC) techniques were performed for vimentin, desmin, cytokeratins, smooth muscle actin, epithelial membrane antigens, IgG4/IgG and ALK (Figs. 2A and B, Figs. 3A and B). Translocation for the ALK gene was also analyzed using the FISH technique (Fig. 3C). The definitive diagnosis of IPT or IMT was based on histopathological, immunohistochemical, and cytogenetic criteria. Four cases were classified as IMT and 9 as IPT. The median tumor size measured in the resection specimen was 14 mm (range: 9–85 mm). The 4 patients diagnosed with IMT were positive for ALK, while the 9 diagnosed with IPT showed IgG4 expression in associated plasma cells (IgG4/IgG ratio), this being one of the diagnostic criteria followed. The main differences between IPT and IMT in our series are listed in Table 1.
 H-E: proliferation of fibroblasts with no signs of malignant disease and chronic inflammation, interspersed with a predominance of plasma cells; B) Immunohistochemistry: positive plasma cells for IgG4 (yellow arrows).")
 H-E: fibrohistiocytic cell proliferation, which is organized in short or storiform fascicles, accompanied by prominent inflammatory infiltrate. No necrosis or vascular permeation is observed; B) IHC: immunohistochemistry stain positive for ALK; C) FISH: translocation for ALK gene (yellow arrows).")
A) H-E: fibrohistiocytic cell proliferation, which is organized in short or storiform fascicles, accompanied by prominent inflammatory infiltrate. No necrosis or vascular permeation is observed; B) IHC: immunohistochemistry stain positive for ALK; C) FISH: translocation for ALK gene (yellow arrows).
Characteristics of the series.
| IPT | IMT | |
|---|---|---|
| Age at presentation (years) | 60.1 | 47.8 |
| Sex | ||
| Males | 6 | 2 |
| Females | 3 | 2 |
| Comorbidities | ||
| Tobacco consumption | 4 | 2 |
| Cancer history | 2 | — |
| Clinical manifestations | ||
| Dyspnea | — | 1 |
| Hemoptysis | 1 | — |
| Mean SUVmax (mg/dL) | 4.3 | 2.6 |
| Surgical approach | ||
| VATS | 6 | 2 |
| Thoracotomy | 2 | 2 |
| Sternotomy | 1 | — |
| Surgical technique | ||
| Lobectomy | 4 | 2 |
| Atypical segmentectomy | 4 | 2 |
| Pneumonectomy | 1 | — |
| Median tumor size (mm)a | 17 | 14 |
| Immunohistochemistry | ||
| IgG4 | 9 | — |
| ALK-1 | — | 4 |
IPT: inflammatory pseudotumor; IMT: inflammatory myofibroblastic tumor; VATS: video-assisted thoracoscopic surgery.
After a median follow-up of 49 months, no signs of locoregional or distant recurrence were observed in the patients studied. Only one patient diagnosed with IPT died during follow-up due to an unrelated cause.
DiscussionIPT generally affects older or elderly adults with a mean age of 60–65 years, showing a predominance in men (60%–80%)13, as we observed in our patients. Meanwhile, some studies have shown that IMT more frequently affects children and young adults14,15. However, our series showed a significantly higher mean age (53.5 years). This is explained by the fact that our service does not treat the pediatric population, with the sole exception of the aforementioned case that required our urgent collaboration. Furthermore, we treat a large portion of a rural population with an above-average mean age, which, together with the indolent nature of the symptoms, could explain why our mean age is higher than reports published in the literature. In any case, the small sample size does not allow conclusions to be drawn in this regard.
The clinical presentation of both tumors depends on the location of the lesion and is usually nonspecific (cough, dyspnea, fever, and chest pain). Regarding IPT, Zen et al.16 found that 53% of patients with pulmonary or pleural involvement had no symptoms at the time of diagnosis, and these patients had been diagnosed incidentally. In our series, more than 80% of patients with IPT were asymptomatic. Some 15%–30% of IMT can present as a constitutional syndrome, with thrombocytosis, polyclonal hyperglobulinemia, elevated erythrocyte sedimentation rate, and growth retardation in children9. Out of the 4 patients diagnosed with IMT in our series, only one presented symptoms (hemoptysis), and the preoperative blood test was strictly normal.
The confirmation diagnosis of these tumors is performed by histopathological characterization. In our series and for all cases of both IPT and IMT, the diagnosis was reached through pathological study of the resection piece after surgery. The suggested cut-off point for the histopathological diagnosis of IPT is the presence of more than 50 IgG4-positive plasma cells per high-power field, IgG4/IgG ratio >0.48,17 and serum IgG4 levels greater than or equal to 135 mg/dL18, although the latter may be altered in other diseases and in up to 5% of the healthy population.
Although preoperative determination of IgG4 may be of interest in screening for this disease, the non-specific nature of the symptoms and imaging tests mean that it does not seem to be a cost-effective diagnostic method, except in cases with a high index of suspicion. In addition, 50%–70% of IMT present translocation in the ALK gene on the short arm of chromosome 2 (2p23) that results in aberrant expression of the ALK protein in myofibroblasts, which can be detected by immunohistochemistry or fluorescent in situ hybridization (FISH)19–21. ALK positivity could serve as an exclusion criterion for IPT. However, ALK negativity does not rule out IMT since, as mentioned above, in less than half of IMT, ALK can be negative in IHC or FISH.
Several studies have shown the presence of positive plasma cells for IgG4 in the lymphoid accumulations of IPT, indicating that this type of cells could be related to its pathogenesis3,22,23. Likewise, a chromosomal rearrangement has been described that is present in more than half of IMT8,9,24. These findings support that IMT and IPT should be considered 2 separate diseases, with different pathogenesis and histological/molecular characteristics.
The natural course of both diseases is unknown since there are no long series published regarding the disease or its treatment. In the case of IPT, the goal of treatment is to prevent fibrosis and irreversible organ damage. Initial therapy with corticosteroids has been shown to be effective for both pulmonary and extrapulmonary disease25,26. The same is true for treatment with immunosuppressants27, including rituximab for cases resistant to corticosteroids and other immunosuppressants28,29. In our cases, no corticosteroid therapy was applied prior to surgery because no preoperative diagnosis was available in any patient. Although reports of spontaneous resolution have been published30, many authors recommend surgery if the patient does not present contraindications for both diagnosis and treatment, showing 5-year and 10-year survival rates in patients with complete resections of 96% and 90%, respectively24,31. Our cases have presented zero mortality without recurrence, as all of them had complete resection.
With regards to IMT, in the past it was classified as a benign lesion, although it is currently considered a neoplasm of intermediate biological potential due to its ability to recur and produce metastasis3,32,33. In our series, however, it had a more favorable biological behavior, but the small number of cases makes it necessary to be cautious in this analysis. In any case, surgery is the first-choice treatment in IMT10,34, with a recurrence rate of less than 2% after complete resection. Even so, no differences were observed in cases of incomplete resection9, and the 10-year recurrence-free period was 80%35. Surgery is also the treatment of choice in recurrences, as long as there are no contraindications36.
In conclusion, IPT and IMT are rare lesions with different pathogenesis as well as clinical and histological characteristics. Problems may arise when making a differential diagnosis with other lung neoplasms since the diagnosis is based on specific pathological techniques. Surgery has both a diagnostic and therapeutic role; after complete resection, good survival rates and a recurrence-free period are achieved.
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: Carrasco Rodríguez R, García Fontán EM, Blanco Ramos M, Magdalena Benavides LJ, Otero Lozano D, Moldes Rodriguez M, et al. Seudotumor inflamatorio y tumor miofibroblástico inflamatorio. Criterios diagnósticos y diferencias pronósticas. Cir Esp. 2022;100:329–335.
