




The aim of this study was to analyse prognostic factors for survival and recurrence in patients with resected pancreatic neuroendocrine tumours (PNT).
MethodsMedical records of 95 patients with resected PNT were retrospectively reviewed. The variables studied were: age, sex, form of presentation (sporadic/familial tumours), functionality, type of tumour, localisation, type of surgery, tumour size, multifocal tumours and recurrent rate. The new WHO classification (2010) was used.
ResultsThere were 45 men and 50 women. Mean age was 46.8 years. Regarding the mode of presentation, it was sporadic in 66 patients (69.8%) and 29 cases were familial neuroendocrine tumours (30.2%) in association with MEN 1 syndrome. The 59% of patients suffered from non-functional tumours and 41% were functional: 20 insulinoma, 16 gastrinoma, and 3 glucagonoma. Distal pancreatectomy was the most common surgical procedure, followed by tumour enucleation in 19 patients. According to the WHO classification, 59 patients had a PNT G1, 24 PNT G2 and 12 with a poorly-differentiated carcinoma, respectively. The 5-year survival in well-differentiated tumours was 100%, regardless of the functionality. Sporadic PNT are more commonly unifocal (P<.001), associating liver metastasis. Survival and recurrence rates after a mean follow-up of 85.3 months were 65.8% and 24%, respectively.
ConclusionsIn our experience, WHO classification was an independent prognostic factor in PNT survival.
El objetivo de este estudio fue analizar los factores pronósticos que influyen en la supervivencia y en la recidiva en una serie de pacientes diagnosticados de tumores neuroendocrinos de páncreas (TNEP) y tratados mediante resección quirúrgica.
MétodosSerie retrospectiva de 95 pacientes intervenidos y resecados de TNEP. Las variables estudiadas son: edad, sexo, forma de presentación (esporádica/familiar), funcionalidad, tipo de tumour, localización, cirugía realizada, tamaño tumoral, multifocalidad, tasa de curación y de recidiva. Se ha utilizado la nueva clasificación de la OMS en 2010.
ResultadosDe los 95 pacientes, 45 eran varones y 50 mujeres, con una edad media de 47,6 años. Presentación esporádica en 66 pacientes (69,8%) y familiar en los 29 restantes (30,2%), asociados a síndrome MEN 1. El 59% (56 pacientes) eran no funcionantes y el 41% restante funcionantes. Los TNEP funcionantes incluían 20 insulinomas, 16 gastrinomas y 3 glucagonomas. La técnica quirúrgica más utilizada (42 pacientes) fue la pancreatectomía corporocaudal. Según la clasificación de la OMS (2010), 59 pacientes presentaban un TNEP G1, 24 un TNEP G2 y los 12 pacientes restantes un carcinoma pobremente diferenciado. La supervivencia a los 5 años en los tumores bien diferenciados ha sido del 100%, independientemente de la funcionalidad. Los TNE esporádicos suelen ser unifocales (p < 0,001) y se asocian a metástasis hepáticas. El seguimiento medio ha sido de 85,3 meses, con una tasa de supervivencia del 65,8% y de recidiva del 24%.
ConclusionesEn nuestra experiencia, la clasificación de la OMS (2010) es un factor pronóstico independiente en la supervivencia de los TNEP.
Pancreatic neuroendocrine tumours (PNET) are uncommon neoplasms with a low annual incidence, which is between 0.32/100000 in the US1 and 1.01/100000 in Japan.2 They represent between 1% and 2% of malignant primary pancreatic tumours, although their prevalence in necropsy studies ranges between 0.5% and 10%.3–5 Since the description of carcinoid tumours by Oberndorfer in 1907,6 neuroendocrine tumours have received several names: carcinoid tumours, apudomas and tumours of the diffuse endocrine system.7,8 Currently, they comprise a group of neoplasms within a larger group known as gastroenteropancreatic neuroendocrine tumours.9–12 Symptoms depend on whether or not the tumours are hormone producers (gastrin, insulin, somatostatin, etc.), and when they do not produce hormones they are known as non-functioning PNET. These latter tumours represent 30%–65% of all PNET.13,14 Recently, in 2010, the WHO proposed a new PNET classification based on tumour proliferation and morphology.15 Generally, PNET are slow-growing tumours with better prognosis than pancreatic duct tumours, even in cases of metastasis. The best treatment is surgery when these masses are resectable.16,17 When unresectable, new molecules (everolimus, sunitinib) have been developed to prolong survival in these patients.18,19
The aim of this study is to analyse the prognostic factors that influence survival and recurrence in a series of 95 patients diagnosed with PNET and treated with surgical resection.
MethodsWe conducted a retrospective study of 130 patients diagnosed with PNET over the course of 22 years. Twenty-four patients were considered inoperable due to their tumour extension, and 11 patients had unresectable disease due to invasion of the celiac trunk and superior mesenteric artery or peritoneal carcinomatosis. The remaining 95 patients who underwent surgery and resection are the focus of our study. The cases that presented signs, symptoms and hormonal analytical alterations (known as functioning tumours [FT]) were classified according to the predominating hormonal alteration. Those with no biochemically established hormone alteration or specific clinical syndrome were classified as non-functioning tumours (NFT). As for the presentation type, PNET can appear isolated, unassociated with other diseases, or as part of a MEN 1 syndrome. In our series, which is a retrospective study, the patients included in the study with PNET associated with familial MEN 1 syndrome were previously diagnosed with this syndrome using genetic testing for the MEN 1 gene. This gene is associated with the chromosome 11q13 region and is a suppressor gene containing 10 exons. The genetic study was conducted with genomic DNA obtained from peripheral blood samples from the subjects and family members. The MEN 1 gene test involved sequencing of exons 2–10 of the MEN1 gene and the corresponding intron-exon junctions. Hepatic metastases were either synchronous or metachronous. Furthermore, we used the 2010 WHO tumour classification15 to classify the PNET as: (a) G1, or low grade; (b) G2, or moderate grade; and (c) G3, or poorly differentiated carcinoma. In order to analyse the prognostic factors that influence survival and the recurrence of PNET, we studied the following variables: age, sex, presentation type (sporadic/familial), tumour function, location, tumour size, metastasis, type of surgery performed and 2010 WHO classification grade.
The following analyses were done according to the diagnosed hormonal alterations: (1) In insulinomas, serum levels of glycemia below 50mg/ld. were pathological. In addition, serum levels of insulin and peptide C were obtained at baseline and after a fasting test; (2) In gastrinomas, serum levels of gastrin were studied at baseline and after stimulation with secretin, along with gastric pH levels; (3) In vipomas, serum levels of VIP were determined, and in glucagonomas serum levels of glucagon were defined, while in somatostinomas serum levels of somatostatin and in vipomas serum levels of pancreatic polypeptide were defined. Other analyses included: 5-hydroxyindoleacetic acid in urine, chromogranin A in blood, and tumour markers CEA, Ca 19.9 and Ca 125.
In our series, abdominal ultrasound was conducted in 68.4% of the cases (65 patients) and CT scan was used in the 95 patients. MRI was performed in 27.3% of the cases (26 patients) and endoscopic ultrasound in 34.7% (33 patients). Using imaging tests, FNA samples were taken from the tumours in 24.2% (23 cases). Last of all, PET/CT was done in 29.4% of the cases (28 patients), while an octreotide scan was used in only 23% (9 patients).
Statistical AnalysisThe statistical software used was IBM SPSS (Statistical Package for Social Sciences Inc, Chicago, Illinois, USA), version 19.0 for Mac.20 We conducted a descriptive analysis, a contingency table analysis using the chi-squared test complemented with the residual analysis, a Student's t test and a logistic regression analysis. Survival curves were calculated with the Kaplan–Meier method, and the log-rank test was used to analyse their differences. A P<.05 was considered statistically significant.
ResultsIn our series, 56 of the 95 cases were NFT (59%) and the remaining 39 were FT (41%), 20 of which were insulinomas, 16 gastrinomas and 3 glucagonomas. Mean age was 47.6±16.3 years (range 17–81), and there was no statistically significant difference between FT and NFT. As for sex, 45 were men (47.3%) and 50 were women (52.7%). In the FT, 19 were males and 20 females, while in NFT, 24 were males (42.9%) and 32 females (57.1%). In our series, 66 cases (69.8%) were sporadic forms and the 29 remaining cases (30.2%) had a familial presentation associated MEN 1 syndrome.
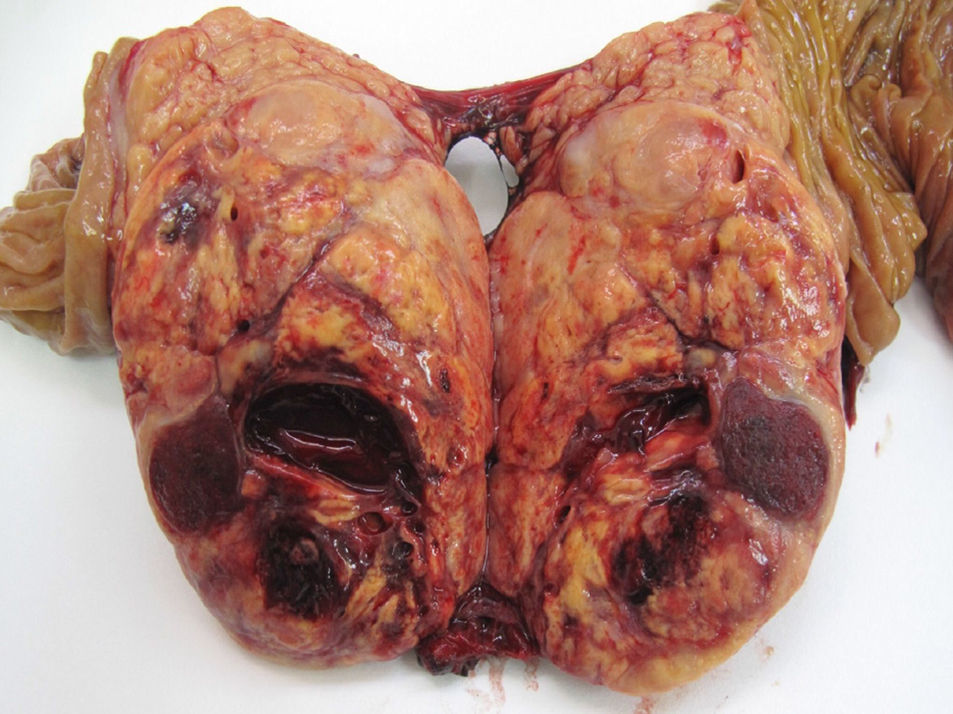
With regard to location, 51.5% of the cases were in the body and tail of the pancreas (49 cases), in 30 patients (31.7%) the location was multifocal, and in 16 patients (16.8%) the location was in the head of the pancreas. Out of the 39 FT, 20 cases were located in the body and tail, 11 cases were multifocal and the remaining 8 in the head of the pancreas. Out of the 56 NFT, 29 cases were located in the body and tail, 19 cases had multifocal distribution and the remaining 8 were in the head of the pancreas. 72.7% (48 cases) of the sporadic forms were solitary, which was statistically significant compared to the familial type (P<.001). Mean tumour size was 3cm (0.4–11cm). If we distinguish between functioning and non-functioning tumours, the mean FT size was 2.4cm (range: 0.4–8cm), versus 3.5cm in the NFT (range: 1.2–11cm), with statistically significant differences (P<.001). With regards to histology grade, 59 cases were G1 or low-grade PNET (62.1%) (Fig. 1), 24 cases were G2 or moderate-grade PNET (25.2%), and in 12 cases the PNET were G3 or poorly differentiated carcinoma (12.6%).
 located in the head of the pancreas.")
In our series, the following surgical techniques were used: (1) distal pancreatectomy in 42 cases (44.2% of patients); (2) pancreaticoduodenectomy in 13 cases (13.7%); (3) tumour enucleation in 19 patients (20%), 12 of which (12.6%) were treated with bilateral subcostal laparotomy and the 7 remaining (7.3%) patients using a laparoscopic approach; (4) distal pancreatectomy+enucleation in 14 cases (14.7%); and (5) total pancreaticoduodenectomy in 7 patients (7.3%). In addition, 3 out of the 7 cases (3.1%) underwent associated portal vein resection with end-to-end anastomosis (2 cases), and a Gore-Tex® graft was inserted in the remaining case. As for the treatment of hepatic metastases (17 cases), liver resection was performed in 7 cases (7.3% of the series) and liver transplantation was conducted in the remaining 10 cases (10.5% of the series). The 10 patients had low-grade sporadic tumours that could not be resected as there were multiple metastases in both liver lobes. Three out of the 10 patients had recurrence from 18 to 24 months post-transplantation and were treated with somatostatin analogues.
In our series, 36 out of the 95 patients (37.9%) presented complications, 25 of which were NFT (26.3% of the series) and the remaining 11 were FT (11.6% of the series). The most frequent were: infection of the surgical wound in 11 cases (11.6%), postoperative abdominal haemorrhage in 10 cases (10.5%) and pancreatic fistula in 8 cases (2 out of 13 pancreaticoduodenectomies and 6 of the 42 distal pancreatectomies). According to the Dindo-Clavien classification,21 8 cases were grade I, 10 cases grade II, 12 cases grade III (5 IIIA and 7 IIIB), 5 cases of grade IV and the remaining case grade V. As for postoperative mortality, only one patient of the non-functioning PNET group died due to abdominal sepsis secondary to pancreatic fistula.
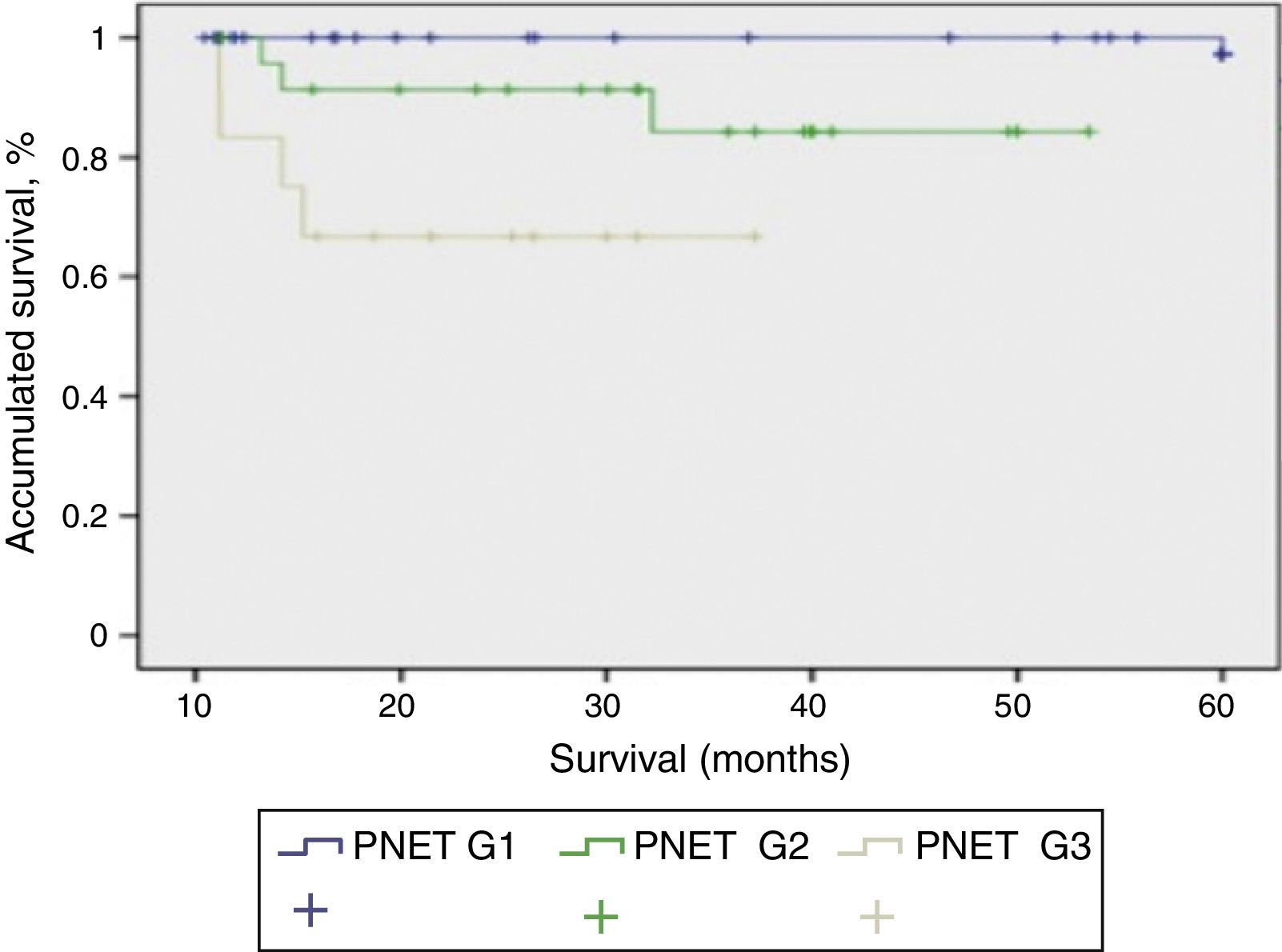
Mean patient follow-up was 85.3 months (range: 4–240 months). During this time, the survival rate was 65.8%, mortality rate was 34.2%, and the tumour recurrence rate was 24%. When we studied the correlation between functionality and presentation type, we found that the patients with sporadic presentation (66 patients) were functioning in 23 cases and non-functioning in 43 cases. Meanwhile, in those with familial presentation, they were non-functioning in 13 cases and functioning in 16 cases. When we used the 2010 WHO classification to analyse survival, we were able to determine the survival of the patients depending on their tumour differentiation grade, with statistically significant differences among the 3 curves (P<.001). In Fig. 2, the G1 PNET showed a 5-year survival rate of 100%. In the G2 PNET, 30-month survival was 90% and 5-year survival was 85%. The worst prognosis is observed in the G3 PNET, or poorly differentiated carcinomas, whose 40-month survival was 65%.
; PNET G1 vs PNET G3 (P<.0001); PNET G2 vs PNET G3 (P<.001).")
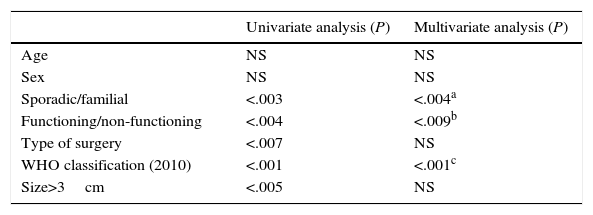
As for the prognostic factors for survival (Table 1), neither age nor sex influenced survival. Nevertheless, the presentation type (sporadic or familial) was a prognostic factor for survival, as the cases with sporadic presentation were associated with poorer prognosis than the familial cases (P<.004, odds ratio [OR] 1.53, 95% CI: 0.87–2.69). Also, tumour functionality (functioning/non-functioning) was a determining factor for survival (P<.001; OR 1.91; 95% CI: 0.95–3.84). In contrast, the type of surgery done and mean tumour size were statistically significant values in the univariate study, while in the multivariate study they were not significant prognostic factors. The 2010 WHO classification was a determinant prognostic factor for PNET survival both in the univariate as well as the multivariate study. The lower the tumour differentiation grade, the worse the prognosis was (P<.001; OR 0.59; 95% CI: 0.48–0.74).
Prognostic Factors for Survival in a Series of 95 Resected Patients.
| Univariate analysis (P) | Multivariate analysis (P) | |
|---|---|---|
| Age | NS | NS |
| Sex | NS | NS |
| Sporadic/familial | <.003 | <.004a |
| Functioning/non-functioning | <.004 | <.009b |
| Type of surgery | <.007 | NS |
| WHO classification (2010) | <.001 | <.001c |
| Size>3cm | <.005 | NS |
CI: confidence interval; NS: not significant; OR: odds ratio.
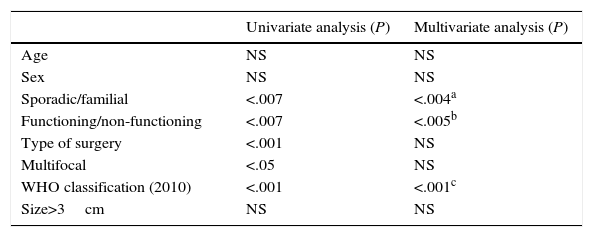
When we analysed the prognostic factors for tumour recurrence (Table 2), age and sex were not statistically significant. Surgery type and multifocal appearance were not significant in the multivariate analyses, although they were significant in the univariate analysis. Only the sporadic or familial type of presentation (P<.004; OR 1.42; 95% CI: 1.08–1.87), tumour functionality (P<.005; OR 0.66; 95% CI: 0.46–0.93) and the 2010 WHO classification (P<.001; OR 1.61: 95% CI: 1.20–2.17) presented statistically significant values in the univariate and multivariate analyses and were prognostic factors for tumour recurrence.
Prognostic Factors for Recurrence in a Series of 95 Resected Patients.
| Univariate analysis (P) | Multivariate analysis (P) | |
|---|---|---|
| Age | NS | NS |
| Sex | NS | NS |
| Sporadic/familial | <.007 | <.004a |
| Functioning/non-functioning | <.007 | <.005b |
| Type of surgery | <.001 | NS |
| Multifocal | <.05 | NS |
| WHO classification (2010) | <.001 | <.001c |
| Size>3cm | NS | NS |
CI: confidence interval; NS: not significant; OR: odds ratio.
PNET are very uncommon neoplasms that originate in the cells of the islets of Langerhans. They may appear at any moment of life, although the maximum incidence is between the 4th and 5th decades.1,5,12 However, when associated with a MEN 1 syndrome, they generally appear before the age of 40.22,23 In our series, mean age was 47.6, and there was a slight predominance of females (52.7%) over males (47.3%), which coincides with other published series.4,5,11 The presentation of PNET is usually isolated, but in 10%–15% of cases they are associated with MEN 1 syndrome, von Hippel-Lindau syndrome, type 1 neurofibromatosis or tuberous sclerosis.22–24 80%–90% of patients with MEN 1 syndrome will develop PNET at some point in their lives, 60% of which will be FT (generally gastrinomas and insulinomas) and the remaining 40% will be NFT.22,25,26 In our series, the PNET were sporadic in 69.8% of the cases and associated with MEN 1 syndrome in the remaining 30.2%.
The use of imaging tests (CT, MRI, etc.) with increasing sensitivity has meant that PNET have been diagnosed more frequently in recent years.27–29 Thoeni et al.27 indicate that, in their series of 28 patients with suspected PNET, MRI had a sensitivity of 85% and a specificity of 100%. Dromain et al.,28 in a series of 64 patients who presented liver metastasis due to PNET, indicated that MRI showed a greater number of hepatic lesions than CT or octreotide scans. Manfredi et al.29 correlated the results obtained with MRI and the biological behaviour of NFT, establishing a relationship between malignancy and the hyperintensity and vascularisation of the lesion in T2. In their article, they report a sensitivity of 93.3% to detect tumours with malignant behaviour. Other techniques, such as endoscopic ultrasound (EUS), have been shown to be useful for detecting lesions of even 2–3mm. Rosch et al.30 and Khashab et al.31 found a greater sensitivity of EUS for detecting this type of tumours versus CT, as well as for the detection and localisation of PNET in patients with MEN 1 syndrome.32 For most authors,30–35 EUS is a highly sensitive test in the detection of PNET that also enables specialists to collect biopsies for histological diagnosis. In our series, it has been used systematically since it was first implemented at our hospital and has demonstrated its utility for locating and determining the size of the lesion as well as for taking biopsies for preoperative histological studies. Octreotide scan or somatostatin receptor scintigraphy may also be useful in the diagnosis of FT, although insulinomas, which are more frequent, express limited levels of somatostatin receptors. Furthermore, octreotide scan can be useful to detect distant metastasis.36–38 PET/CT with 18F-dihydroxyphenylalanine has a greater sensitivity (90%–97%) for detecting and staging PNET39–42 than scintigraphy, and it allows us to detect smaller lesions. In our series, PET/CT was done in 29.5% of the series.
If the tumour is resectable, the treatment of choice is surgery.43–45 The type of surgery depends on the tumour size and location within the pancreatic gland. Thus, in well-defined and small tumours, the procedure involves tumour enucleation; meanwhile, in tumours that present multifocal distribution, total pancreatectomy should be performed. Recently, the laparoscopic approach has been proposed, especially in tumours smaller than 3cm located in the tail of the pancreas.46–48 A meta-analysis by Drymousis et al.,49 including 11 articles with a total of 906 cases, reported that 22% were treated with laparoscopic surgery and 78% with open surgery. After comparing surgical times, hospital stay, intraoperative blood loss, postoperative morbidity, pancreatic fistula and mortality rates, they stated that laparoscopic surgery is a safe technique in the hand of experts and that it is associated with a lower rate of complications and shorter hospital stay than open surgery. In our series, 52% of the cases were located in the body and tail, 32% were multifocal, and the remaining 16% were located in the head of the pancreas. According to tumour resectability and location, the most frequent surgical techniques were distal pancreatectomy in 42 cases and enucleation in 19 cases. The approach was laparoscopic in 7.3% of cases.
In PNET, metastases are usually located in the liver. The capacity for metastatic extension is determined by the histologic type and the proliferative rate of the tumour. Furthermore, they have a peculiar biological behaviour, as the metastatic disease is usually confined to the liver for long periods with 5-year survival rates, even without being treated, of around 30%.50,51 Treatment is determined by the number of metastases and their distribution. Surgical techniques involve liver resection or liver transplantation. Liver resection is the treatment of choice for curative intent, as long as there is no diffuse involvement, decline in liver function or extrahepatic metastasis. In a series of 170 patients who had been treated with liver resection due to hepatic metastases of PNET, Sarmiento et al.52 reported a long-term disease-free survival of 20%. In this series, there was clinical improvement in 90% of the cases, although the recurrence rate was 84% and 5-year and 10-year survival rates were 61% and 35%, respectively. As for liver transplantation, Lehnert et al.53 stated that this should be done in young, symptomatic patients with poor response to pharmacological treatment and in absence of extrahepatic disease. Gedaly et al.54 published a report by the United Network for Organ Sharing (UNOS) in which 150 liver transplantations were carried out in patients with neuroendocrine tumours and hepatic metastatic disease from 1988 to 2008. In this series, the 1-year, 3-year and 5-year survival rates of the patients were 81%, 65% and 49%, respectively. These survival rates were comparable to those of a group of 4693 patients who had received transplants for hepatocellular carcinoma during the same period of time. However, the majority of the patients with neuroendocrine tumours treated with liver transplantation presented disease recurrence.55–57 In our series, hepatic metastases were detected in 17 cases: in 7 cases liver resection was performed, and in 10 patients liver transplantation was necessary.
The prognosis of PNET is difficult to determine as it is equally difficult to define the benign or malignant nature of the disease. Several PNET classifications have been described, although the most widely used is the WHO classification from 2010.15 Based on this classification, 62.1% of the cases (59 patients) in our series were well-differentiated endocrine tumours, 25.2% (24 cases) were well-differentiated endocrine carcinomas and the remaining 12.6% (12 cases) were poorly-differentiated endocrine carcinomas. Some authors58 consider that in NFT there is a close relationship between tumour size and risk for malignancy, which increases for tumours larger than 2cm. Bilimoria et al.,59 in a series of 3851 patients and after a multivariate analysis, considered that age, tumour grade and metastases are the most significant factors that influence survival.
Generally, 5-year and 10-year survival rates for PNET is 65% and 45%, respectively.60 In FT, 5-year survival is 80%, versus 55% of NFT. For several authors,61–63 radical resection of the primary tumour, the presence of liver metastases and their treatment are factors that influence the survival of patients with PNET. Yang et al.64 indicated that the histological type, Ki-67 proliferation index, size, location, patient age, TNM classification and the 2010 WHO classification are prognostic factors that influence survival of patients with PNET.
In our series, poorly differentiated tumours had a poorer prognosis than well-differentiated tumours, as they had a 5-year survival lower than 65% while the 5-year survival was 100% in well-differentiated tumours. Moreover, the rate of tumour recurrence was higher in poorly differentiated tumours. We also found that sporadic tumours presented shorter survival rates than PNET associated with MEN 1 syndrome. This is because sporadic presentations are more frequently NFT than familial presentations; meanwhile, their diagnosis is delayed and with a greater probability for extension of the disease.
To conclude, our experience demonstrates that the prognostic factors for tumour recurrence and survival include presentation type, tumour function and the 2010 WHO classification grade, as the lower the degree of tumour differentiation, the poorer the prognosis.
Authorship/collaborations- 1.
Study design: Sánchez Bueno, Francisco; Rodríguez González, José Manuel; Torres Salmerón, Gloria; and Parrilla Paricio, Pascual.
- 2.
Data collection: Sánchez Bueno, Francisco; Rodríguez González, José Manuel; Torres Salmerón, Gloria; Bernabé Peñalver, Antonio; and Balsalobre Salmeron, María.
- 3.
Analysis and interpretation of the results: Sánchez Bueno, Francisco; Rodríguez González, José Manuel; Balsalobre Salmerón, María; de la Peña Moral, Jesús; and Fuster Quiñonero, Matilde.
- 4.
Article composition: Sánchez Bueno, Francisco; Rodríguez González, José Manuel; Torres Salmerón, Gloria; and Bernabé Peñalver, Antonio.
- 5.
Critical review and approval of the final version: Sánchez Bueno, Francisco; Rodríguez González, José Manuel; and Parrilla Paricio, Pascual.
The authors have no conflict of interests to declare.
The authors would like to thank Dr. María Antonia Claver from the Nuclear Medicine Department and Dr. Eduardo Ortiz from the Pathology Department.
Please cite this article as: Sánchez-Bueno F, Rodríguez González JM, Torres Salmerón G, Bernabé Peñalver A, Balsalobre Salmeron M, de la Peña Moral J, et al. Factores pronósticos de los tumores neuroendocrinos de páncreas resecados. Experiencia en 95 pacientes. Cir Esp. 2016;94:473–480.
