




The calcifying cystic fibrous tumour (CCFT) is an extremely rare benign tumour first described in 1998 in children, as a childhood fibrous tumour with psammoma bodies.1 Subsequently, in 1993 a series of 10 cases was described in patients between 1 and 33 years of age, and was named calcifying fibrous pseudotumour.2 At present, the WHO recommends calling it a tumour, due to the possibility of local recurrence.3
We present the case of a 31-year-old woman with grade II obesity associated with arthritis, high percentage of body fat, glucose intolerance, hyperfibrinogenemia, hyperandrogenism and hyperleptinemia. During surgery–laparoscopic tubular gastrectomy–multiple implants were observed mainly in the greater omentum with a pearly appearance (between 1 and 4mm) of hard consistency, and encapsulated, suggestive of peritoneal carcinomatosis. An intraoperative biopsy was performed and the result was inconclusive; although the signs were compatible with a benign process a peritoneal dissemination due to a digestive or gynaecological tumour could not be ruled out.
Before surgery the patient had had a gynaecological examination and a colonoscopy, and both had been normal. Furthermore, during evaluation for bariatric surgery, an upper endoscopy had been performed that was also normal.
The abdominal cavity, pelvis, Douglas cul-de-sac, both lower quadrants, ovaries, liver and spleen were carefully inspected, and no pathologic findings were found. Only one other mass of a larger size–1cm–but of similar characteristics, was found close to the cecal appendix. Because of the uncertainty of diagnosis, the bariatric procedure was postponed, and omentectomy and appendectomy were performed with a therapeutic intent, as the appendix was considered as the possible primary tumour, in order to obtain an R0 resection and await the definitive pathological diagnosis.
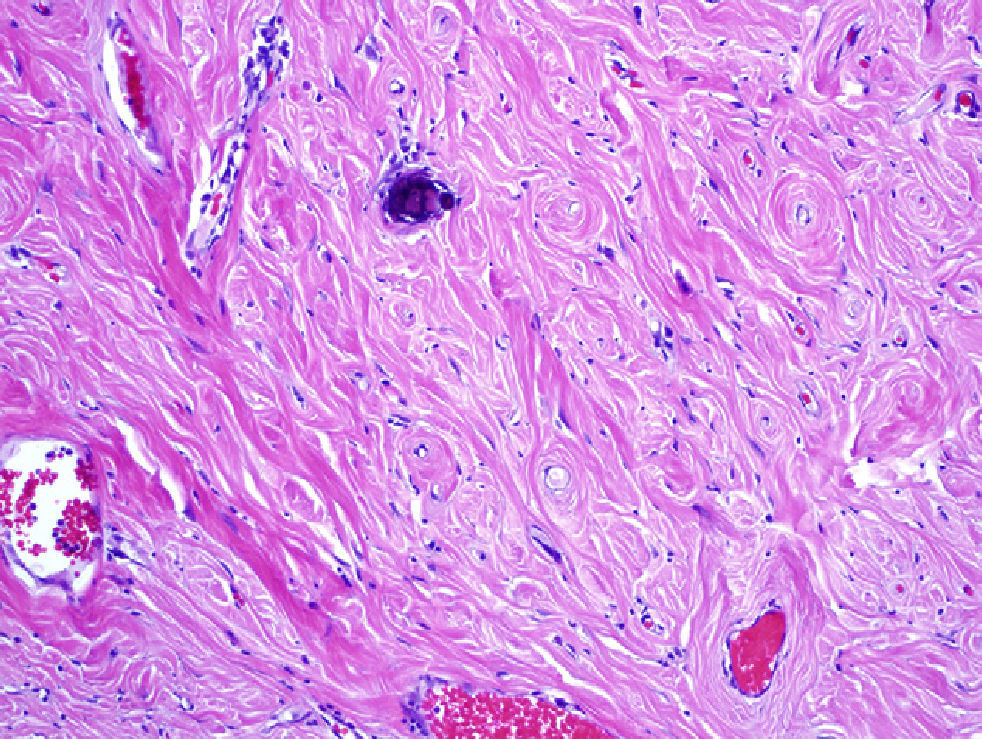
The pathology study showed nodules formed by thick collagen bundles with hyalinised areas, some of them calcified with psammmoma bodies. A vascular network of fine walls with an inflammatory infiltrate surrounding it predominantly of plasmocytes and occasional lymphocyte was observed. The nodules were well delimited from the fibroadipose tissue and covered by a layer of mesothelium with focal hyperplasia in small nests (Fig. 1). An immunohistochemical analysis was performed for antibodies against CD-32, CD-34, Keratin AE3/AE1, cytokeratin 5/6 and calretinin, and a diagnosis of multiple peritoneal CCFT was made.

CCFT is an exceptionally rare benign tumour; to date there have only been 70 cases reported and the published series are very small.3,4 It is considered that 90% are solitary tumours located in soft tissues: suprarenal glands, spleen, liver, stomach, spermatic chord, pleura, peritoneum or omentum.4,5 Only 10% of CCFT are multiple and there are only 3 cases described of multiple abdominal CCFT,3,5–7 4 if we add the present case, which would also be the first multiple peritoneal CCFT described in the Spanish literature. The clinical presentation of CCFT is variable, from an asymptomatic mass or incidental finding at surgery or autopsy to abdominal pain similar to an acute appendicitis in the second or third decade of life, with no difference between the sexes.4–6,8,9
The aetiology of this tumour is unknown although some authors have related it to chronic inflammatory states, trauma, prior surgery or a late sclerosing stage of an inflammatory myofibroblastic tumour. There are also theories about a certain genetic base and there is a case described of familial presentation.3,5,6,10 Image diagnosis is performed using CT scan, as plain X-rays and ultrasound have a low efficiency.5
The definitive diagnosis is exclusively pathological. Macroscopically they present as well circumscribed tumours in soft tissue areas that can vary in size from a few millimetres to 25cm, and can be solitary or multiple.4,9 Microscopically, they present nests of fibrous tissue and hyalinized collagen, with scarce cellularity, and psammoma bodies.3–7,9,10
The immunohistochemical studies reveal a positive reaction to vimentin, actin 1 A4, and variable to CD 34, and are negative for ALK.3,9,10 Differential diagnosis should be made with a peritoneal carcinomatosis, inflammatory myofibrobastic tumour, nodular fasciculitis, fibromatosis, aponeurotic calcifying fibroma or amyloidoma.9,10
Definitive treatment is surgical resection of the lesion or lesions and a laparoscopic approach is valid, although it has been performed in only two cases including this one.3,9,10 Recurrence is rare and no metastatic spread has been described, therefore an extensive follow-up does not seem necessary.4,6,7,9
Please cite this article as: Bellver Oliver M, Arredondo Chaves J, Queipo Gutierrez P, Valentí Azcárate V, Rotellar Sastre F. Tumor fibroso quístico calcificante. Una extraña forma de carcinomatosis peritoneal benigna. Cir Esp. 2013;91:338–339.
