




El tumor fibroso quístico calcificante (TFQC) es un tumor benigno excepcional de partes blandas descrito por primera vez en 1988 en niños, como tumor fibroso de la infancia con cuerpos de Psamoma1. Posteriormente, en 1993 se describió una serie de 10 casos en pacientes de entre 1 y 33 años, siendo denominado entonces como pseudotumor fibroso calcificante2. Actualmente la OMS recomienda llamarlo tumor dada su capacidad de recurrencia local3.
Presentamos el caso de una mujer de 31 años de edad con obesidad grado II que asociaba sobrecarga articular, porcentaje de grasa corporal muy elevado, intolerancia a la glucosa, hiperfibrinogenemia, hiperandrogenismo e hiperleptinemia. Durante la cirugía -gastrectomía tubular laparoscópica- se visualizaron sobre todo el epiplón mayor múltiples implantes milimétricos de aspecto nacarado (entre 1 y 4mm) de consistencia dura, aspecto encapsulado que eran sugestivos de una carcinomatosis peritoneal por lo que se decidió realizar una biopsia intraoperatoria cuyo resultado no fue concluyente, ya que pese a presentar signos muy sugestivos de benignidad no se podía descartar diseminación peritoneal de un tumor de origen digestivo o ginecológico.
Antes de la intervención la paciente había realizado una revisión ginecológica y una colonoscopia, ambas sin hallazgos patológicos. Además, dentro del preoperatorio de cirugía bariátrica, se había realizado una gastroscopia previa que resultó compatible con la normalidad.
Ante esta situación se inspeccionó detalladamente la cavidad abdominal, pelvis, fondo de saco de Douglas, ambas fosas ilíacas, ovarios, hígado y bazo, sin hallazgo patológico. Tan solo se localizó una masa de mayor tamaño -1cm-, pero de características similares, próxima al apéndice cecal. Dada la situación de incertidumbre, se decidió posponer la cirugía bariátrica hasta la obtención de un resultado concluyente, se realizó omentectomía y apendicectomía con intención terapéutica al considerar el apéndice un posible foco primario, en cuyo caso se habría conseguido una resección R0, en espera de un diagnóstico anatomopatológico definitivo que permitiera tomar una actitud terapéutica correcta.
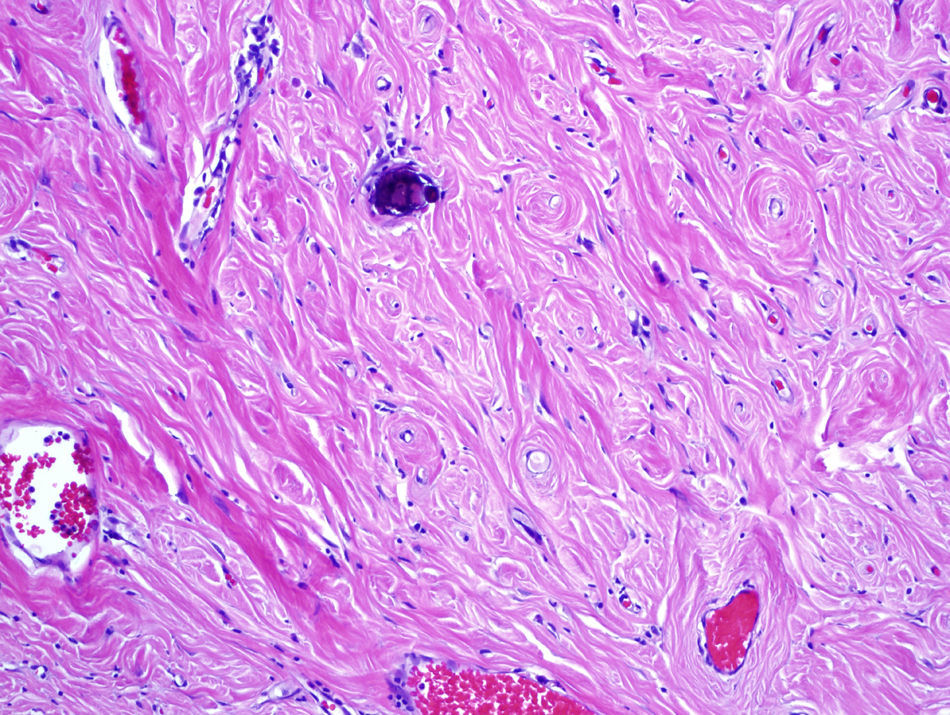
La anatomía patológica informó de nódulos constituidos por haces de colágeno gruesos con áreas hialinizadas, con calcificaciones en algunos de ellos en forma de cuerpos de Psamoma. Destacaba una red vascular de pared fina, en torno a la que se disponía un infiltrado inflamatorio predominantemente plasmocitario y muy ocasionalmente con linfocitos. Los nódulos estaban bien delimitados con respecto al tejido fibroadiposo adyacente y recubiertos por una capa de mesotelio con hiperplasia focal en pequeños nidos. (fig. 1.) Tras realizar un estudio inmunohistoquímico utilizando anticuerpos frente a CD-31, CD-34, queratina AE3/AE1, citoqueratina 5/6 y calretinina, se diagnosticó como TFQC múltiple peritoneal.

El TFQC es un tumor benigno excepcionalmente raro del que hasta la fecha no se conocen más de 70 casos y las series descritas son de muy pocos casos3,4. Se calcula que el 90% son tumores únicos localizados en tejidos blandos: suprarrenales, bazo, hígado, estómago, cordón espermático, pleura, peritoneo o epiplon4,5. Tan solo el 10% de los TFQC son múltiples y únicamente hay 3 casos descritos de TFQC múltiple abdominal3,5–7, 4 si sumamos el que presentamos, que además supondría el primer caso de CFT múltiple peritoneal descrito en la literatura española. La presentación clínica del TFQC varía desde una masa indolora o un hallazgo incidental en una cirugía o en autopsias hasta un dolor abdominal similar a una apendicitis entre la segunda y tercera décadas de la vida, sin predilección especial por el sexo4–6,8,9.
La etiología de este tumor es desconocida aunque algunos autores lo han relacionado con estados inflamatorios crónicos, traumatismos, cirugías previas o un estadio tardío esclerosante del tumor inflamatorio miofibroblástico. También existen teorías sobre cierta base genética e incluso se ha descrito un caso de agregación familiar3,5,6,10. El diagnóstico por imagen queda prácticamente reducido a la TC, puesto que la radiología simple o la ecografía ofrecen un escaso rendimiento5.
El diagnóstico definitivo es exclusivamente anatomopatológico. Macroscópicamente se presentan como tumores circunscritos en regiones de tejidos blandos que oscilan desde los pocos milímetros hasta los 25cm, pudiendo ser únicos o múltiples4,9. Microscópicamente llama la atención la presencia de nidos de tejido fibroso y colágeno hialinizado, de escasa celularidad, con cuerpos de Psamoma en su interior3–7,9,10.
En la inmunohistoquímica destaca la reactividad positiva para vimentina, actina 1 A4 y variable para CD 34, siendo negativa para ALK3,9,10. Su diagnóstico diferencial debe hacerse con una carcinomatosis peritoneal, tumor inflamatorio miofibroblástico, fasciculitis nodular, fibromatosis, fibroma calcificante aponeurótico o amiloidoma9,10.
El tratamiento definitivo es la resección quirúrgica de la lesión o lesiones siendo válido el abordaje laparoscópico, realizado en solo dos ocasiones incluyendo este caso3–7,9,10. La recidiva es excepcional y nunca se han descrito metástasis, por lo que no parece necesario un seguimiento exhaustivo4,6,7,9.

