




There is no doubt about the relationship between LDL-C and cardiovascular risk, as well as about the benefits of statin treatment. Once the objective of LDL-C has been achieved, the evidences that demonstrate the persistence of a high cardiovascular risk, a concept called residual risk, are notable.
The residual risk of lipid origin is based on atherogenic dyslipidemia, characterised by an increase in triglycerides and triglyceride-rich lipoproteins, a decrease in HDL-C and qualitative alterations in LDL particles. The most commonly used measures to identify this dyslipidemia are based on the determination of total cholesterol, triglycerides, HDL, non-HDL cholesterol and remaining cholesterol, as well as apolipoprotein B100 and lipoprotein(a) in certain cases.
The treatment of atherogenic dyslipidemia is based on weight loss and physical exercise. Regarding pharmacological treatment, we have no evidence of cardiovascular benefit with drugs aimed at lowering triglycerides and HDL-C, fenofibrate seems to be effective in situations of atherogenic dyslipidemia.
Es indudable la relación del cLDL y el riesgo cardiovascular, así como de los beneficios del tratamiento con estatinas. Una vez conseguido el objetivo de cLDL, son notables las evidencias que demuestran la persistencia de un elevado riesgo cardiovascular, concepto denominado riesgo residual.
El riesgo residual de origen lipídico se fundamenta en la dislipidemia aterogénica, caracterizada por un aumento de triglicéridos y de las lipoproteínas ricas en triglicéridos, un descenso del cHDL y alteraciones cualitativas de las partículas LDL. Las medidas más utilizadas para identificar esta dislipidemia se basan en la determinación de colesterol total, triglicéridos, HDL, colesterol no HDL y colesterol remanente, además de las apolipoproteínas B100 y la lipoproteína(a) en determinados casos.
El tratamiento de la dislipidemia aterogénica se basa en la pérdida de peso y ejercicio físico. En cuanto al tratamiento farmacológico, no tenemos evidencia del beneficio cardiovascular con los fármacos dirigidos al descenso de triglicéridos y cHDL; el fenofibrato parece tener eficacia en situaciones de dislipidemia aterogénica.
Current thinking is that the priority in reducing cardiovascular risk (CVR) is to lower cholesterol bound to low-density lipoproteins (LDL-C). This criterion is based on extensive epidemiological evidence and prospective interventional studies in primary and secondary prevention which demonstrate that a 25–40% reduction in LDL-C is associated with a 9–38% decrease in CVR, based on the initial level of that risk.1–11 Several meta-analyses based on prospective epidemiological studies have shown a continuous linear relationship between LDL-C concentrations and the risk of atherosclerotic vascular disease. The Emerging Risk Factors Collaboration, using data from 302,430 individuals with no cardiovascular disease (CVD) at the time of inclusion and from 68 prospective studies, confirmed that plasma LDL-C concentration was linearly associated with an increased risk of myocardial infarction or cardiovascular mortality.12 Similarly, the Prospective Studies Collaboration, in a meta-analysis of 892,337 individuals with no CVD included in the study from 61 prospective cohort studies, confirmed a clear linear association between cholesterolaemia and the risk of coronary artery disease-related mortality.13
LDL cholesterol as a fundamental part of cardiovascular riskStatin therapy has been shown to be clearly effective in the primary and secondary prevention of CVD, and statins are in fact the drugs of choice for lowering LDL-C. In the Cholesterol Treatment Trialists’ (CTT) Collaboration, in a meta-analysis of 26 interventional studies with statins and nearly 170,000 individuals, statin therapy was associated with a linear reduction of cardiovascular episodes.14 In this meta-analysis, the absolute reduction in LDL-C was directly proportional to the reduction in primary vascular episodes: the more marked the reduction in LDL-C, the greater the benefit. For each 1mmol/l (39mg/dl) reduction in LDL-C, the risk of vascular episodes was reduced by approximately 20%, regardless of the initial cholesterol concentration, which implies that an additional reduction of 2–3mmol/l would reduce the risk by approximately 40–50%. These findings revealed that the primary objective for patients with high CVR should be to achieve the greatest possible reduction in LDL-C without increasing the risk of myopathy, regardless of the target figures proposed at the time. Data from this meta-analysis were confirmed by the SEARCH study, which compared the use of simvastatin 80mg/day vs. 20mg/day in over 12,000 secondary prevention patients over seven years. Supporting this was the finding that the greatest reductions in LDL-C were associated with a greater reduction in CVR.15
The reduction in LDL-C in patients with type 2 diabetes mellitus (T2DM) is of particular significance in reducing their high CVR. In the Collaborative AtoRvastatin Diabetes Study (CARDS) conducted in 2838 diabetic patients with no previous cardiovascular history, LDL-C was reduced by 40% and the incidence of cardiovascular episodes by 37% in a follow-up period of up to nine years.16 The CTT study confirmed the benefit of reducing LDL-C with statins in diabetic patients, with a 21% decrease in cardiovascular episodes per mmol/l of LDL-C reduction.17 A subgroup of nearly 5000 diabetic patients from the IMPROVE-IT study confirmed that the combination of simvastatin and ezetimibe, in addition to achieving a greater reduction in LDL-C (0.4mmol/l), reduced cardiovascular episodes by 14%.18 However, it should be noted that, despite the cardiovascular benefits of using maximum doses of statins,19 the CVR in patients with T2DM remains high (Table 1) and life expectancy is lower.11
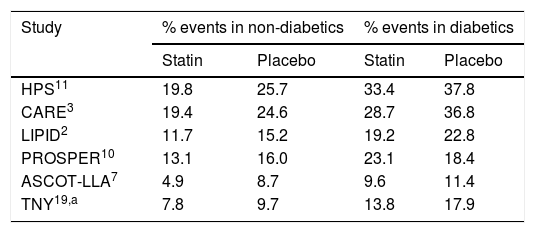
Various studies confirm that there is still a high residual cardiovascular risk in patients with T2DM following statin therapy.
| Study | % events in non-diabetics | % events in diabetics | ||
|---|---|---|---|---|
| Statin | Placebo | Statin | Placebo | |
| HPS11 | 19.8 | 25.7 | 33.4 | 37.8 |
| CARE3 | 19.4 | 24.6 | 28.7 | 36.8 |
| LIPID2 | 11.7 | 15.2 | 19.2 | 22.8 |
| PROSPER10 | 13.1 | 16.0 | 23.1 | 18.4 |
| ASCOT-LLA7 | 4.9 | 8.7 | 9.6 | 11.4 |
| TNY19,a | 7.8 | 9.7 | 13.8 | 17.9 |
In all the studies, cardiovascular death and non-fatal myocardial infarction were evaluated.
In a recent meta-analysis designed to evaluate the prevention of severe vascular episodes with various lipid-lowering treatments, involving 19 clinical trials and 152,507 secondary prevention patients with a mean follow-up of four years,20 it was confirmed that the reduction in relative risk was more significant the higher the initial LDL-C concentration or the greater its reduction. Each mmol/l reduction in LDL-C led to a 19% reduction in the relative risk of severe cardiovascular episodes. The three most important conclusions of this meta-analysis are as follows: the most intensive treatment showed a significant reduction in mortality (cardiovascular and all-cause), the reduction in cardiovascular episodes is proportional to the reduction in LDL-C, and intensification of treatment with non-statin drugs is associated with significant mortality reductions in very high-risk patients.
The new lipid-lowering treatment based on proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, used as monotherapy and in combination with statins, reduces LDL-C concentration by approximately 45–60%. The FOURIER study included 27,564 patients with CVD and LDL-C levels >70mg/dl who received statin therapy and who were randomised to evolocumab or placebo for 48 weeks.21 The study drug arm reduced LDL-C by 59%, with an absolute difference of 53.4mg/dl compared to the placebo group. After a mean follow-up of 2.2 years, the primary endpoint (cardiovascular mortality, myocardial infarction, stroke, coronary artery bypass or hospitalisation due to unstable angina) fell by 15% and the risk was reduced by 11% for each 38.6mg/dl reduction in LDL-C. The ODISSEY OUTCOMES study22 included 18,924 patients from 57 countries who had suffered a myocardial infarction or unstable angina during the previous 12 months. Patients were selected on the basis of one of the following criteria: LDL-C ≥70mg/dl, non-HDL cholesterol ≥100mg/dl or apolipoprotein (Apo) B ≥80mg/dl. Following intensive statin therapy for 2–16 weeks, patients were randomised to receive alirocumab or placebo. After a follow-up period of two to five years (mean 2.8 years), the recently published results23 demonstrated that LDL-C levels were 53.3mg/dl in the alirocumab group compared to 101.4mg/dl in the placebo group (absolute reduction of 54.7%). The primary endpoint of major cardiovascular events was significantly lower in the alirocumab group than in the placebo group (9.5 vs. 11.1%). More specifically, with the study drug, non-fatal myocardial infarction was reduced by 14%, stroke by 27% and unstable angina by 39%. In the post hoc analysis by LDL-C level at the start of the study, patients with LDL-C ≥100mg/dl experienced greater reductions in all evaluation criteria.
We do not know a great deal about the effect of PCSK9 inhibitors on other lipid fractions with atherogenic potential that could affect CVR. However, there is one recent meta-analysis with pooled data from ten phase 3 studies belonging to the ODYSSEY study in which the effect of alirocumab in patients with metabolic syndrome was analysed: Apo B in these patients decreased from 35 to 55% and levels of cholesterol not bound to high-density lipoproteins (non-HDL-C) decreased from 38 to 54%, percentages similar to those seen in patients without metabolic syndrome.24 In another recent meta-analysis with evolocumab25 involving patients with T2DM, the reduction in non-HDL-C was 55% vs. ezetimibe and 34% vs. placebo. In the ODYSSEY LONG TERM study,26 non-HDL-C decreased 51.6% vs. 0.7% with placebo and Apo B decreased 52.8% vs. 1.2% with placebo. In the ODYSSEY DM-DYSLIPIDEMIA study, however, performed in patients with T2DM and dyslipidaemia (non-HDL-C ≥100mg/dl and triglycerides [TG] ≥100mg/dl), patients were randomised to receive alirocumab or, in addition to therapy with the maximum tolerated dose of statins, other commonly used lipid-lowering drugs (fenofibrate, ezetimibe, omega-3, nicotinic acid or diet alone). In this study, alirocumab reduced LDL-C (43%), Apo B (32.3%) and the number of LDL particles (37.8%) compared to the usual selected treatment (p<0.0001).27 According to these data, we can conclude that PCSK9 inhibitors have a clear and significant action on the group of atherogenic lipoproteins.
Residual risk independent of LDL cholesterolThe aforementioned data are unquestionably clear about the effect of LDL-C on CVR: the more marked the reduction of this lipoprotein, the greater the reduction in CVR. However, despite obtaining LDL-C values within the range of the targets established by various scientific societies and despite even achieving the lowest possible values with statins and non-statin drugs, there is still a high, unacceptable and proven risk of cardiovascular disease. Moreover, there is still a high percentage of cardiovascular episodes, especially in secondary prevention patients. This CVR, which persists despite treatment, is largely due to lipid factors other than LDL-C that have a potentially atherogenic action,28 hence the name residual CVR of lipid origin.
This residual risk of lipid origin reflects aspects related to triglyceride-rich lipoproteins (TGRL), which includes in particular very low-density lipoproteins (VLDL). Enzymatic hydrolysis of VLDL leads to the formation of smaller and denser particles which are rich in cholesterol. Experimental studies show that these lipoproteins could contribute to the development of CVD,29,30 a concept which has been adopted by various consensus panels and scientific societies.31,32
Furthermore, some epidemiological studies have highlighted the significance of the residual risk of lipid origin in total CVR. In the REsiduAl risk, LIpids and Standard Therapies (REALIST) study, it was confirmed that TG and HDL-C are closely related to residual risk in patients with acute coronary syndrome and with LDL-C between 70 and 130mg/dl. Furthermore, higher TG concentrations associated with lower HDL-C concentrations increase the risk of coronary heart disease tenfold. In the Prospective Cardiovascular Münster study, the CVR for acute myocardial infarction increases fivefold in patients with LDL-C within the normal range, but with an increased TG or decreased HDL-C.33
In view of these findings, it is important to identify other lipid factors besides LDL-C which are associated with the residual risk of lipid origin, not only in order to correct them but also to identify them in patients with a high CVR, who frequently present with these lipid changes, as occurs in situations of insulin resistance such as T2DM, obesity or metabolic syndrome.
The body of evidence available has supported the significance of atherogenicity of TGRL and their remnants, often in combination with low HDL-C concentrations, and currently they are even considered secondary therapeutic targets in high/very high CVR patients. The Residual Risk Reduction Initiative (R3i) has highlighted the importance of this dyslipidaemia as a contributing factor in lipid-related residual CVR.34 In fact, a post hoc analysis of trials with fibrates showed that individuals with this dyslipidaemic profile had a relative reduction of 35% in the risk of cardiovascular episodes, compared to 6% in those subjects without dyslipidaemia.35
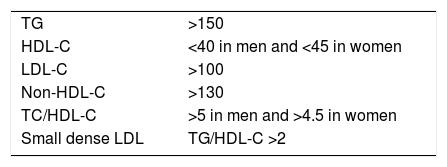
The combination of increased TGRL, reduced HDL-C and increased small, dense LDL particles has led to the concept of atherogenic dyslipidaemia (AD), which is largely responsible for the residual risk of lipid origin (Table 2). If the prevalence of metabolic syndrome is approximately 22–39% of the adult population, and the lipid components of this syndrome coincide with those of AD, we could estimate the high prevalence of this lipid disorder in the general population. We will analyse the components of AD and the measures required for its diagnosis below.
Atherogenic dyslipidaemia.
| TG | >150 |
| HDL-C | <40 in men and <45 in women |
| LDL-C | >100 |
| Non-HDL-C | >130 |
| TC/HDL-C | >5 in men and >4.5 in women |
| Small dense LDL | TG/HDL-C >2 |
In mg/dl.
Source: Ascaso et al.77
At present there is sufficient scientific evidence for hypertriglyceridaemia to be considered an independent risk factor for CVD.36 A meta-analysis which analysed the association between TG and CVD in the general population, based on prospective studies with a total of 65,863 men and 11,089 women, revealed that an increase of 89mg/dl in TG concentration was associated with a 12% increase of CVR in men and a 37% increase in women, after adjusting for total cholesterol, LDL-C, HDL-C, body mass index, blood pressure and diabetes.37
Elevated TG are independently associated with T2DM, metabolic syndrome, obesity and chronic kidney disease.38 Moreover, this increase is affected by diet, certain drugs, genetic factors and, in particular, lifestyle. It is estimated that up to a quarter of the US adult population has hypertriglyceridaemia, defined as TG>150mg/dl.39 Severe hypertriglyceridaemia, defined by the National Lipid Association guidelines as TG>500mg/dl, is a clear risk factor for acute pancreatitis.40 However, it has been shown that moderately elevated TG are independently associated with a greater risk of CVD, even in patients effectively treated with statins to reduce LDL-C.41,42
The Emerging Risk Factors Collaboration study, carried out in the general population with no previous CVD, revealed that triglyceridaemia was associated with various cardiovascular episodes, although this association disappeared following adjustment for HDL-C and non-HDL-C.43 In the Copenhagen General Population Study, a strong association was also observed between postprandial TG and CVR.44 During the follow-up of the Bezafibrate Infarction Prevention study, mortality data were analysed after a period of more than 20 years of follow-up and in over 15,000 secondary prevention patients, and it was proven that the risk of mortality at 22 years increased by 68% in patients with hypertriglyceridaemia compared with patients with low or normal TG (p<0.001). Therefore, elevated TG in patients with CVD are associated with a higher risk of long-term mortality, regardless of HDL-C. Severe hypertriglyceridaemia (TG>500mg/dl) identifies a population with an especially high risk of mortality.45
In the PROVE-IT TIMI study, conducted in patients with acute coronary syndrome receiving treatment with statins and target LDL-C (<70mg/dl), a TG concentration >150mg/dl was independently associated with a lower rate of recurrence.39 The pooled analysis of the IDEAL and TNT studies also demonstrated that, in secondary prevention patients treated with statins, a decrease in TG was accompanied by a reduction in the incidence of cardiovascular episodes, although this association diminished after adjustment for HDL-C.46
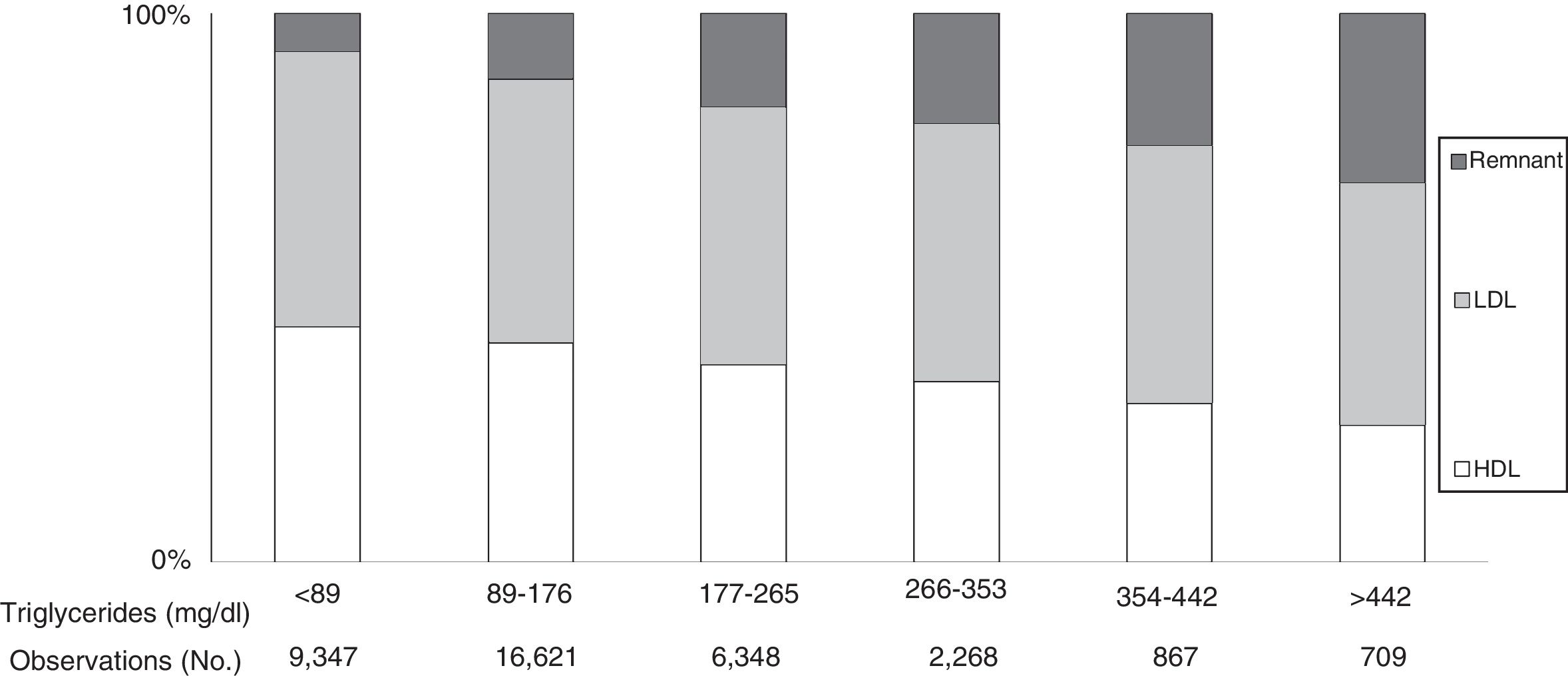
Plasma TG levels are known to correspond to levels of TGRL and their remnants (Fig. 1).31 However, hypertriglyceridaemia is also usually accompanied by other lipoprotein changes, such as elevated VLDL and Apo C-III, a predominance of small, dense LDL particles and reduced HDL-C levels, all closely related to a greater risk of CVD.47
 increased, while LDL concentrations showed no changes and HDL levels tended to decrease. Under these conditions, remnant cholesterol represents the total cholesterol transported in IDL, VLDL and chylomicron remnants. Source: Modified from Chapman et al.31")
Cholesterol in lipoproteins according to elevated triglyceride levels in the general population. Based on the Copenhagen general population study. 9% of men and 6% of women took statins. As triglyceride levels increased, remnant lipoproteins (VLDL, IDL and remnant) increased, while LDL concentrations showed no changes and HDL levels tended to decrease. Under these conditions, remnant cholesterol represents the total cholesterol transported in IDL, VLDL and chylomicron remnants. Source: Modified from Chapman et al.31
The fact that the results of studies with TG-lowering drugs have produced inconsistent cardiovascular outcomes48 illustrates that there is still a certain degree of uncertainty regarding the direct causal role of TGRL in the progression of atherosclerosis and CVD.
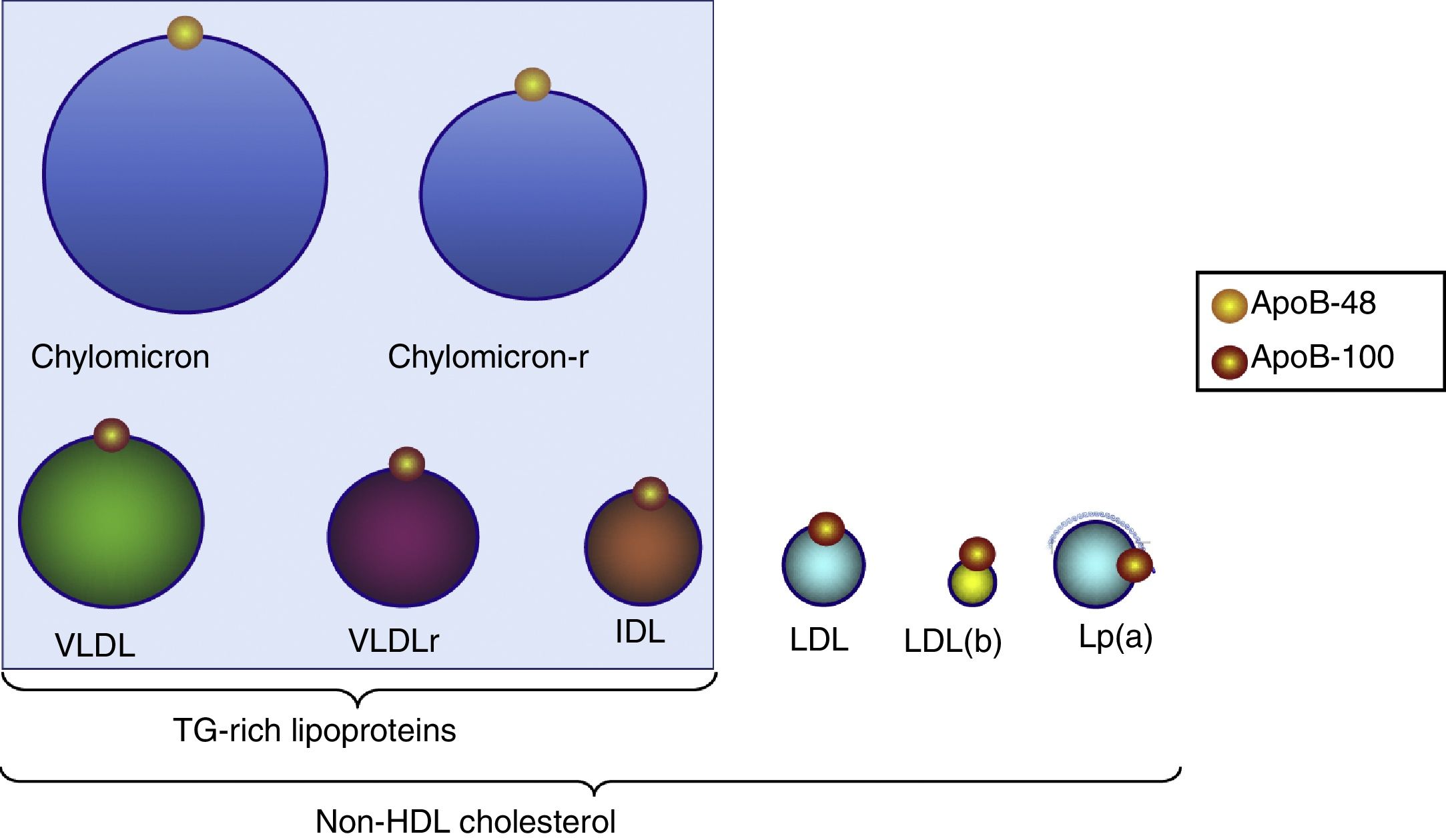
Triglyceride-rich lipoproteinsTG are transported in plasma by VLDL, chylomicrons and their remains which originate during their metabolism (remnants). TGRL are the largest lipoprotein particles and are characterised by the apolipoproteins they contain, such as apoB-100, which is associated with VLDL and intermediate-density lipoproteins [IDL] (Fig. 2) and apoB-48, which is associated with chylomicrons and their remnants.
: small, dense LDL; chylomicron-r: residual chylomicron or remnants; VLDLr: residual VLDL or remnants.")
Distribution of triglyceride-rich lipoproteins and particles included in non-HDL-C. All these lipoproteins contain one apoB-100 molecule and, therefore, measurement of this apoprotein indicates the number of particles with atherogenic potential. LDL(b): small, dense LDL; chylomicron-r: residual chylomicron or remnants; VLDLr: residual VLDL or remnants.
TGRL are highly heterogeneous due to their different size, density and composition. They are composed of a neutral lipid core (consisting of TG and cholesteryl esters) and a surface monolayer of phospholipids, free cholesterol and the apolipoproteins which participate in the regulation of transport and their metabolism.49
It has been suggested that it is primarily the cholesterol content of the TGRL, rather than the TG themselves, that contributes directly to the progression of atherosclerosis.50 Like LDL, TGRL which are low in TG and rich in cholesterol can penetrate the arterial intima and be trapped in the subendothelial space, where they undergo phagocytosis by macrophages, which contributes to foam cell formation and the formation and progression of atherosclerotic plaque. It is accepted that TGRL are equal to or even more atherogenic than LDL, since, in contrast to these, the remnant particles of TGRL can be absorbed directly by the macrophages without oxidative modification. In addition, because of their greater size, they transport more cholesterol per particle than LDL.51 It has also been demonstrated that TGRL remnants promote endothelial dysfunction, which potentiates atherogenesis.52
Lipoprotein lipase-mediated hydrolysis of TGRL results in a high concentration of free fatty acids in the endothelium or even within the arterial intima. These lipolytic products, together with TGRL themselves, activate several pro-inflammatory, pro-coagulant and pro-apoptotic signalling pathways, which play a fundamental role in the pathogenesis of atherosclerosis. We know that oxidised free fatty acids increase the expression of inflammatory interleukins and cytokines, which leads to endothelial inflammation,53 while TGRL remnants facilitate the expression of endothelial adhesion molecules,54 which are pro-atherogenic, by facilitating the transendothelial migration of leukocytes and the activation of monocytes and neutrophils.55
However, TGRL remnants increase the production of reactive oxygen species with a consequent increase in endothelial permeability. They also promote leucocyte adhesion and increase the secretion of pro-apoptotic cytokines, tumour necrosis factor α and interleukin-1β, which leads to vascular injury and the development of the atherosclerotic process.56
TGRL and their remnant particles increase platelet aggregation and cause coagulation cascade amplification through the prothrombinase complex and increased expression of plasminogen activator inhibitor-1, in addition to encouraging tissue factor expression in endothelial cells.57,58 Finally, there is evidence that TGRL remnants suppress the atheroprotective and anti-inflammatory effects of HDL59 and it has been demonstrated that they are significantly correlated with the deterioration of coronary vasodilation.60 Mendelian randomisation studies, in which the genes involved in TGRL metabolic pathways (Apo A5, Apo C3) were used, have demonstrated that these lipoproteins are causal factors of CVD.
Remnant cholesterolResidual or remnant cholesterol is the cholesterol contained in TGRL, i.e. in VLDL, in IDL and in chylomicron remnants. It can be calculated simply by subtracting LDL-C and HDL-C from total cholesterol. The difference with respect to non-HDL-C is that this contains residual cholesterol plus LDL-C. The significance of remnant cholesterol is that it must be identified in subjects with normal or low LDL-C, since in these cases elevated values can occur which would not be identified by evaluating non-HDL-C alone. Current thinking is that remnant cholesterol is an important factor in CVR.50,61
Data are available demonstrating the association between high concentrations of remnant cholesterol and the elevated risk of coronary artery disease,62 myocardial infarction63 and all-cause mortality.64 In the recently published Copenhagen General Population Study conducted in a total of 106,216 patients who were followed-up for 11 years, subjects were distributed by quartiles of remnant cholesterol and according to low weight, normal weight, overweight or obesity. The conclusion of this study is that remnant cholesterol is positively correlated with body mass index and that remnant cholesterol is a risk factor for myocardial infarction independent of the anthropometric parameter.65 In a preliminary result from the same study, performed in 90,000 subjects from the general population, it was proven that a progressive increase in remnant cholesterol and LDL-C was associated with an increased risk of coronary artery disease and myocardial infarction. However, although increasing concentrations of remnant cholesterol were associated with higher all-cause mortality, this was not the same for LDL-C. Even after reducing LDL-C to the recommended concentrations, there was still a substantial risk of CVD, and, consequently, the authors concluded that for future interventional studies, it is necessary to reduce both LDL-C and remnant cholesterol.66
Non-HDL cholesterolNon-HDL-C is the sum of LDL, lipoprotein(a) (Lp[a]), VLDL and IDL cholesterol. It is calculated by simply subtracting HDL-C from total cholesterol. At present, it is considered a reliable marker of CVR as it encompasses all the atherogenic lipoproteins containing cholesterol and Apo B. Moreover, it is easy to calculate and does not interfere with other lipid assays. It is a very useful measurement in clinical situations related to insulin resistance, which are very common in T2DM, metabolic syndrome or visceral obesity, in which AD is a characteristic disorder, and, therefore, it may be considered an important marker of residual risk of lipid origin. In these patients, it has been recommended that the most accurate therapeutic target is non-HDL-C, or rather Apo B, since both parameters are better correlated to CVR than LDL-C,67 and there is even evidence that non-HDL-C is superior to LDL-C in evaluating CVR.68
This situation confirms that, although LDL-C may remain more or less stable, as atherogenic remnant lipoproteins increase (lipoproteins containing Apo B) and HDL-C decreases, the risk is increased, which represents all atherogenic cholesterol and not just LDL-C. If in this situation only LDL-C were to be used, we would be underestimating the risk because it would not reflect the real increase in atherogenic lipoproteins.69 In a recent study performed in patients with T2DM and LDL-C <100mg/dl, it was proven that the elevated Apo B/Apo A-I ratio (>0.57) is independently associated with the risk of carotid arteriosclerosis.70
The latest clinical guidelines from the American Association of Clinical Endocrinologists and the American College of Endocrinology clearly show that an increase in non-HDL-C is a greater risk factor with importance equal to that of LDL-C or total cholesterol and equivalent to hypertension, T2DM, chronic kidney disease, smoking or a family history of CVD. These same guidelines set out five risk categories (low, moderate, high, very high and extreme) and set therapeutic targets for non-HDL-C for each of the risk categories mentioned.71 The calculation of non-HDL-C for risk stratification in secondary prevention individuals with moderately high TG (>200mg/dl) and diabetes is also recommended. Moreover, in subjects with suspected insulin resistance (metabolic syndrome), non-HDL-C should be evaluated to consider the atherogenic lipoprotein burden.
A meta-analysis of eight studies with 62,154 patients treated with statins showed that non-HDL-C had a better correlation with CVR than LDL-C and Apo B. Even subjects who achieved only the therapeutic LDL-C targets showed an increased risk of 32% (95% CI: 1.17–1.50) compared with those who achieved both targets.70 In another recent meta-analysis of nine prospective studies involving the general population, which included 448,732 individuals, it was shown that patients with higher non-HDL-C at the start of the study had a higher risk of CVD (RR 1.79; 95% CI: 1.68–1.91) compared with those with the lowest level of non-HDL-C.72
Number of LDL particlesThe subfractions of LDL particles can be determined by various methods (density gradient ultracentrifugation, electrophoresis and chromatography), although the use of nuclear magnetic resonance spectroscopy is currently the most effective method since it provides rapid quantification of the size and concentration of LDL particles and various subclasses of lipoproteins, including VLDL, IDL, LDL and HDL. Consequently, it is a potential and important aid for rigorously evaluating CVR in clinical practice.
Large prospective studies (EPIC-Norfolk, Framingham Offspring, MESA, Women's Health Studies) have demonstrated that the quantification of LDL particles has greater predictive value of CVR than the LDL-C value. This concept is important since many patients with AD (T2DM, metabolic syndrome) have an increased number of LDL particles without showing increased LDL-C. In this situation, patients often have higher TGRL concentrations, which may explain the greater risk of CVD. The number of LDL particles may also be considered a marker of subclinical CVD as they are associated with intima-media thickness and arterial calcium to a larger extent than LDL-C.73
The presence of LDL subclasses depends on the hepatic availability of TG: if it is low, the liver primarily segregates VLDL1 (rich in TG) and IDL. VLDL1 are modified by lipoprotein lipase and hepatic lipase to produce LDL-III, which by means of hepatic lipase and cholesteryl ester transfer protein (CETP) end up forming small dense LDL particles. Starting as IDL, LDL-I is formed through lipoprotein lipase. Conversely, when TG availability is high, a pattern different to secretion takes place: large VLDL1 and VLDL2 are produced, after the action of lipoprotein lipase and hepatic lipase, the first are transformed into LDL-IV and the second into IDL1 and LDL-II, by both pathways and by means of hepatic lipase and CETP small dense LDL are produced.74 In summary, we could assert that an increase in TGRL causes an increase in the exchange of these with cholesteryl esters coming from the various LDL particles through the action of the CETP, and, by means of this enzymatic action, the formation of smaller and denser LDL particles is increased.75
Small dense LDL particles have a lower affinity for the LDL receptor, which slows down their clearance from the circulation. The fact that they remain longer in plasma and are smaller facilitates penetration into the arterial intima and retention in the subendothelial space due to their greater affinity for proteoglycans. Furthermore, these particles are more susceptible to oxidation, which is a determining factor in the process of atherogenesis. This pattern of LDL particles is clearly increased in T2DM and is closely related to the TGRL concentration since it is observed in up to 90% of patients with TG>150mg/dl.
On most occasions, LDL-C concentration and the number of LDL particles are closely related, and therefore plasma LDL-C is a good substitute for the concentration of LDL particles. However, in certain conditions (metabolic syndrome, T2DM and hypertriglyceridaemia), the LDL-C concentration and number of LDL particles may be conflicting on account of the predominance of small, dense LDL particles, and, consequently, the plasma LDL-C may not reflect the LDL particle concentration. Under these conditions, direct measurement of the number of LDL particles or the Apo B concentration (understanding that each LDL particle contains a single molecule of Apo B) may reflect a greater causal effect than LDL itself for atherosclerotic CVD.76
The quantity of these particles may be estimated indirectly by the TG/HDL-C ratio, which, if greater than 2, indicates its increase and the range to be considered in AD.77
It is currently accepted that all LDL particles should be considered atherogenic,78 a finding revealed by patients with familial hypercholesterolaemia, who show a predominance of large LDL particles and premature atherosclerosis.79 However, a review of 24 studies which reported on the relationship between LDL subfractions and cardiovascular events concluded that a greater number of LDL particles, more than the size of these particles, was associated with a greater risk of cardiovascular events.80
HDLHDL are responsible for reverse cholesterol transport, i.e. from the peripheral tissues to the liver, one of the primary anti-atherogenic mechanisms.
HDL particles are multi-molecular, composed predominantly of polar lipids, in addition to proteins (enzymes) and small quantities of non-polar lipids, which are present in multiple isoforms. As a result of their composition, they are highly heterogeneous in their structural, chemical and biological properties. There are two types: HDL2, the less dense particles with a higher lipid content, and HDL3, which are denser and richer in proteins. HDL2 and HDL3 can be fractionated into various subclasses with different electrophoretic mobilities. There are two subclasses of HDL2 and three of HDL3.
Increased TGRL also lead to TG being transferred to HDL by the CETP. The TG enrichment of HDL makes them more susceptible to the action of hepatic lipase, an enzyme which acts on HDL2 particles (greater size, lesser density and variable cholesterol content) and transforms them into smaller HDL3 particles (small, dense, with minimal lipid content), which are rapidly cleared from plasma and eliminated via the kidneys. It has been proven that TG levels modulate the concentration of HDL subclasses, since the increase in TG is accompanied by HDL of a smaller size, particularly due to the decrease of the HDL2 subclass and increase of HDL3. These changes in HDL are associated with a drop in their free cholesterol content and an increase in TG. The result of this process is a net decrease in HDL and an increase in small dense LDL particles. This dyslipidaemia is usually accompanied by an increase in non-HDL-C and, as a result, an increase in Apo B since all non-HDL particles carry this Apo. Elevated TG concentrations and decreased HDL-C are accompanied by a high CVR, even in patients with desirable LDL-C concentrations.81
HDL, in addition to facilitating reverse cholesterol transport, also directly protect the vascular wall by other mechanisms: they reduce cell adhesivity to the endothelium with a resulting reduction in the endothelial migration of monocytes, facilitate the production of prostacyclin (a vasodilator and inhibitor of platelet aggregation), interfere with fibrinolysis and reduce the cellular and body fluid mediators of arterial inflammation.82
Accordingly, the aforementioned Emerging Risk Factors Collaboration study highlighted HDL-C as a predictive risk factor independent of coronary and cerebrovascular episodes, even when it was adjusted for other lipid and non-lipid risk factors. The SPARCL study, in patients with previous cerebrovascular disease, showed a greater decrease in the risk of recurrence of stroke when the HDL-C levels were higher, regardless of LDL-C values.83 The meta-analyses by the CTT in primary and secondary prevention, and in patients treated with statins, indicated unequivocally that, regardless of LDL-C values, CVR is lower with higher HDL-C values, even in cases of lower LDL-C.84,85 Another study in patients with stable coronary artery disease receiving high-intensity statin therapy (TNT) also highlighted the predictive power of low HDL-C concentrations, even with well-controlled LDL-C (<70mg/dl).81 In patients with acute coronary syndrome treated with statins (MIRACL study), it has been observed that HDL-C, and not LDL-C, was a predictor, regardless of short-term prognosis.14 Finally, a meta-analysis of 20 studies comprising a total of 70,939 patients with statins and 66,068 with placebo demonstrated that statins did not change the relationship between HDL-C and CVR. Accordingly, low HDL-C levels remain significantly and independently associated with a greater risk in spite of statin therapy.86 Patients treated with statins had the same level of risk associated with lower HDL-C levels as control subjects (even after adjustment for LDL-C during treatment, age, hypertension, diabetes and tobacco consumption).67 These findings highlight the CVR associated with low HDL-C levels, even in patients treated with statins.
Lipoprotein(a)This lipoprotein is considered an additional and independent marker of CVR.87,88 Lp(a) is similar to LDL. It also contains apoB-100 as well as a specific Apo, Apo(a), which is structurally identical to plasminogen. The plasma Lp(a) concentration is determined genetically. Systematic determination of the plasma Lp(a) concentration is not recommended for detecting risk in the general population. However, Lp(a) measurements should be considered in individuals at high risk of CVD or with a family history of premature cardiovascular episodes. The risk is considered significant when the Lp(a) concentration is above the 80th percentile (50mg/dl).89 Likewise, it has been demonstrated that Lp(a) enables correct reclassification of risks87,90 and should be considered in borderline high- and moderate-risk patients.
Therapeutic approach in residual cardiovascular riskAn appropriate therapeutic approach for AD should have a positive effect on the residual risk of lipid origin, which persists after the effective use of statins for the purpose of reducing LDL-C levels. Consequently, identifying the lipid factors associated with the residual risk can be particularly significant, not only to treat them but also to detect them in patients with various lipid profile changes associated with a high CVR, such as patients with T2DM and characteristic AD in over 50% of the cases.
The various clinical guidelines include non-HDL-C or Apo B as secondary endpoints once the primary endpoint for LDL-C has been achieved, or, in some cases, LDL-C and non-HDL-C are even put forward as endpoints with the same level of importance. In all cases, lifestyle modification (diet and exercise) is crucial for reducing residual risk. Pharmacological strategies for reducing this risk are also available and will be discussed below.
Diet and exerciseA diet rich in monounsaturated fats significantly improves insulin sensitivity, which is associated with a reduction in TG levels, particularly postprandially. A decrease in TG is shown when saturated fats are replaced by omega-6 polyunsaturated fatty acids (PUFA), or with high doses of omega-3 intake. However, a therapeutic approach based only on foods will make it difficult to achieve a significant clinical effect. To achieve this objective, supplements or foods enriched artificially with omega-3 PUFA may be used. In subjects with severe hypertriglyceridaemia, even with the presence of chylomicrons in a fasting state, the total quantity of dietary fat should be reduced as much as possible (<30g/day). In these patients, the use of medium-chain TG (C6 to C12) may be considered, which prevent the formation of chylomicrons since they are directly transported and metabolised in the liver by the hepatic portal system.91
The relationship between carbohydrate and lipid metabolism is also known. Therefore, a high dietary carbohydrate intake may lead to increased TG concentrations.92 This occurs particularly with the intake of fast-acting (refined) carbohydrates with a high glycaemic index. This effect is reduced considerably with the consumption of complex carbohydrates and those with a high-fibre content, and is applicable to patients with T2DM or metabolic syndrome.
Weight reduction improves insulin sensitivity, causing TG levels to decrease and achieving reductions of up to 30%, which are maintained as long as weight is not regained. Carrying out regular physical exercise reduces TG levels regardless of weight loss. Alcohol consumption has a major effect on triglyceridaemia. Whereas in individuals with hypertriglyceridaemia even a small amount of alcohol can induce a greater rise in TG concentrations, among the general population alcohol intake only affects TG levels if consumption is excessive.93
With regard to HDL-C, saturated fatty acid intake increases such levels commensurately with the increase in LDL-C.94 The consumption of mono- or polyunsaturated fat has practically no effect on HDL-C. Increased carbohydrate consumption as a substitute for fat is associated with decreased HDL-C (0.4mg/dl reduction for every 1% replacement), an effect which is not affected by fibre content or the glycaemic index.
Moderate alcohol consumption is associated with higher HDL-C levels compared with non-drinkers. Weight loss has a clear benefit in terms of HDL-C levels: it increases 0.4mg/dl per kilogram of weight loss. Aerobic physical activity, such as fast walking 3.5–4.5km/day (or any equivalent activity), can increase HDL-C levels between 3 and 6mg/dl.95 Quitting smoking can also help increase HDL-C, provided that weight gain, which usually occurs after quitting smoking, is avoided.
DrugsTo date, clinical trials conducted with various drugs aimed at reducing TG and increasing HDL-C, and thereby reducing the residual risk of lipid origin, have had minimal success in reducing CVR.
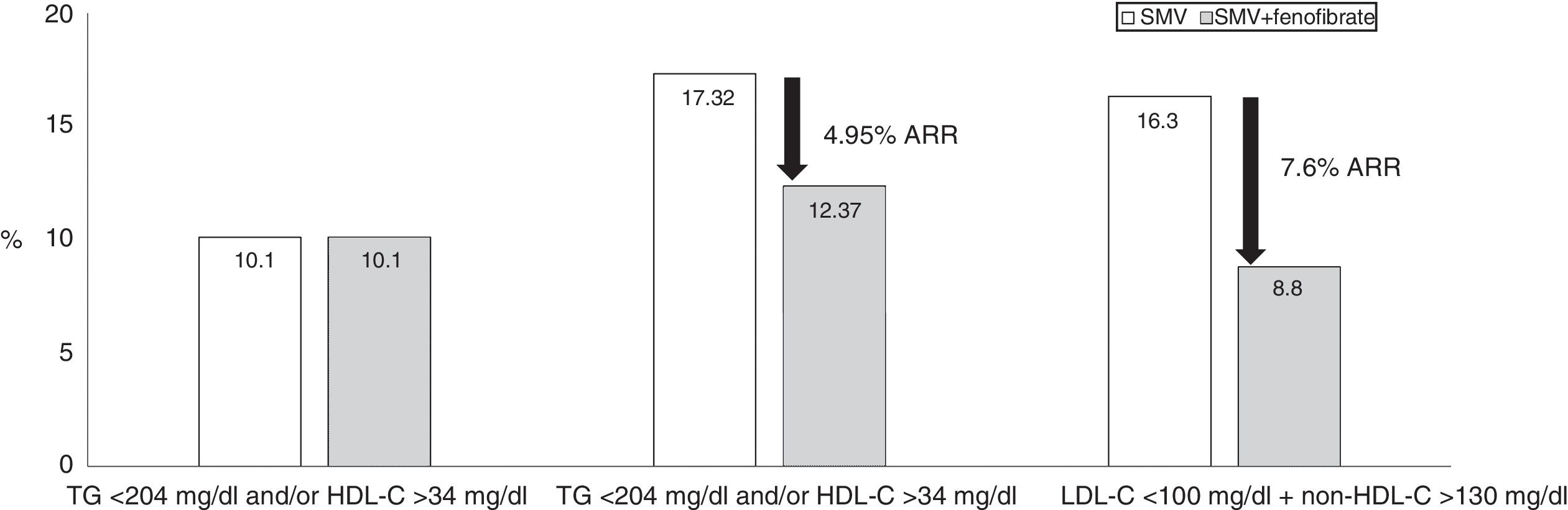
To reduce TG, the drug fenofibrate has demonstrated a certain degree of efficacy on residual risk.96 Furthermore, when combined with statins, the safety of this combination has been sufficiently proven in various clinical trials.97–99 This combination has demonstrated, in addition to reducing the LDL-C values, a clear decrease in TG levels and an increase in HDL-C levels. The combination of simvastatin and fenofibrate in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) study constitutes the first description of the efficacy of this combination, although it did not significantly reduce non-fatal cardiovascular episodes and cardiovascular death in diabetic patients compared to simvastatin monotherapy. However, patients from the AD group showed a 70% higher relative risk of severe cardiovascular episodes compared with diabetic patients without dyslipidaemia, despite achieving a mean LDL-C level of 80mg/dl (Fig. 3). One specific analysis showed that in patients with elevated TG (>200mg/dl) and reduced HDL-C (<34mg/dl), the relative CVR was reduced by 31% in the combined therapy group compared to simvastatin monotherapy.100 To prevent a cardiovascular episode, only 20 patients needed to be treated with combined therapy for five years (absolute risk reduced by 4.95%). The Fenofibrate Intervention and Event Lowering in Diabetes Study was designed to evaluate the long-term effect of fenofibrate on cardiovascular episodes in patients with T2DM.101 A total of 9795 patients aged between 50 and 75 participated in this study. No significant reductions in the primary mortality endpoint occurred, but treatment with fenofibrate reduced total cardiovascular episodes. The patients who benefited the most were those with TG >150mg/dl and HDL-C <40mg/dl in men and 50mg/dl in women. This study also demonstrated a beneficial effect of fenofibrate on microvascular risk, with a reduction in the progression of oligoalbuminuria, the development and progression of retinopathy and the risk of lower extremity amputation. Consequently, to address the reduction of residual risk, particularly in patients with AD in whom it appears necessary to consider optimising the therapeutic results in the lipid triad (LDL-C, HDL-C and TG), it may be worth evaluating the possible combination of statins and fenofibrate, as has been proven to date.102
, but with non-HDL-C >130mg/dl. The major cardiovascular events were: cardiovascular death, non-fatal myocardial infarction or stroke. Source: Modified from The ACCORD Study Group.100")
Accord-lipid study: 5518 patients with T2DM, 4.7 years follow-up. The presence of atherogenic dyslipidaemia increased the risk of major cardiovascular events by 70%. The combination of simvastatin and fenofibrate significantly reduced the risk in subjects with LDL-C within range (<100mg/dl), but with non-HDL-C >130mg/dl. The major cardiovascular events were: cardiovascular death, non-fatal myocardial infarction or stroke. Source: Modified from The ACCORD Study Group.100
Another drug with the ability to reduce TG by up to 25% and increase HDL-C concentrations between 15 and 25%103 is nicotinic acid or niacin, which, when combined with a statin, may offer potential benefits for reducing residual risk of lipid origin104. Patient adherence for this drug is usually irregular due to its side effects, primarily flushing. Nicotinic acid can reduce insulin sensitivity, which encourages the development of T2DM in predisposed subjects or worsens metabolic control in diabetic subjects.105 In the AIM-HIGH study, conducted in patients with atherosclerotic CVD and LDL-C levels of less than 70mg/dl, no clinical benefit was observed with the combination of niacin and statins over a follow-up period of 36 months, despite improvements in HDL-C and TG levels.106 In the HPS2-THRIVE study, conducted in patients with atherosclerotic vascular disease, the addition of extended-release niacin-laropiprant to statin therapy did not significantly reduce the risk of major vascular events, but did increase the risk of severe adverse events.107 In other studies, a 12–14% reduction in coronary mortality108 has been proven with the use of niacin and a statin, and a reduction of carotid intima-media thickness greater than that obtained with the intensification of statin monotherapy109 has been demonstrated. Despite the potential beneficial effects demonstrated in the aforementioned multicentre studies, use of this drug is, at present, practically non-existent due to its side effects.
Finally, another compound indicated for reducing TG is omega-3 fatty acids, administered at a dose of 3–4g/day, which significantly reduce TG and TGRL, both in monotherapy and in combination with a statin.110,111 In secondary prevention, the use of omega-3 fatty acids has shown a reduction in cardiovascular morbidity/mortality112: even a dose of 1g/day reduces mortality and cardiovascular-related hospitalisation in patients with heart failure.113 However, several subsequent meta-analyses have demonstrated that treatment with omega-3 fatty acids is not associated with a protective effect against the main primary cardiovascular events, though it does have beneficial effects in reducing death due to cardiac causes, sudden cardiac death and all-cause mortality.114,115
Finally, pharmacological action to increase HDL-C is represented by CETP inhibitors. Some of the most notable studies conducted with these drugs are those carried out with torcetrapib (ILLUSTRATE, RADIANCE, ILLUMINATE). These three studies, however, had to be discontinued after 18 months due to increasing cardiovascular events by 25% and due to 40% more deaths in the active arm.116 The dal-OUTCOMES study, performed with dalcetrapib combined with statins in patients with acute coronary syndrome, was also stopped because of a lack of efficacy in the active arm.117 The REVEAL study, with anacetrapib, demonstrated cardiovascular benefits from the third year, without reducing mortality.118 Evacetrapib also improved the lipid profile, but did not reduce cardiovascular events compared to placebo.119
Generally, all pharmacological actions intended to reduce residual CVR of lipid origin, having TG or HDL-C as targets, have had minimally beneficial or even contradictory results, except in certain post hoc analyses in specific populations. Accordingly, no current guideline recognises TG and HDL-C as therapeutic endpoints.
ConclusionThere is an enormous amount of scientific evidence available on the effect of LDL-C on CVR, as well as its relationship with cardiovascular morbidity and mortality. However, the concept of residual risk of lipid origin comprises persistent and elevated CVR once the LDL-C objective has been achieved, within the values accepted by the majority of scientific societies. The factors responsible for this residual lipid risk include TG and the lipoproteins which transport them (TGRL), in addition to a reduction of HDL-C and an increase of LDL particles. These lipid changes are defined by the concept of AD. To identify such changes and offer an intervention plan we utilise simple calculations, such as non-HDL-C or remnant cholesterol, which have been incorporated as therapeutic endpoints in most clinical guidelines, particularly the former. Finally, in addition to dietary modifications and increased physical exercise, we have a limited therapeutic arsenal to combat this risk. Fenofibrate is the only drug that is effective in patients with AD. New non-statin drugs related to an increase of LDL receptors appear to be effective, but limited use in routine clinical practice reduces the potential benefit among those with an elevated residual lipid risk.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Hernández-Mijares A, Ascaso JF, Blasco M, Brea Á, Díaz Á, Mantilla T, et al. Riesgo cardiovascular residual de origen lipídico. Componentes y aspectos fisiopatológicos. Clín Investig Arterioscler. 2019;31:75–88.