




Vitamin D deficiency is a worldwide pandemic and results in osteoporosis, hypertension, and other cardiovascular diseases. At the cellular level, it produces significant oxidative stress, inflammatory markers, and mitochondrial damage. There is increasing evidence about the role of vitamin D in the regulation of the renin-angiotensin-aldosterone system (RAAS). Moreover, there is evidence of involvement in cardiovascular complications, as well as in the immune system disorders. Vitamin D values below 25 ng/mL are related to an increase in vascular tone mediated by smooth muscle contraction. Furthermore, it can produce direct effects on vascular smooth muscle cells, RAAS over-regulation, modulation of calcium metabolism, and secondary hyperparathyroidism. All this predisposes patients to develop hypertrophy of the left ventricle and vascular wall, causing hypertension. In this work, a review is presented of the main mechanisms involved in the development of hypertension due to vitamin D deficiency. Among them are the link established between the levels of extra-mitochondrial inorganic phosphate, its main regulatory hormones—such as vitamin D—, the cardiovascular system, reactive oxygen species, and mitochondrial metabolism. The role of the mitochondrial vitamin D receptor and the regulation of the respiratory chain could influence arterial remodelling since its activation would reduce oxidative damage and preserve cell life. However, there are aspects not yet understood about the intricate signalling network that appeared simple in experimental trials, but complex in clinical studies. In this way, the completion of new studies as “VITAL”, could clarify, and thus support or refute the possible benefits of Vitamin D in hypertensive cardiovascular disease.
El déficit de vitamina D es una pandemia a nivel mundial y trae como consecuencia osteoporosis, hipertensión arterial (HTA) y otras enfermedades cardiovasculares. A nivel celular produce mayor carga oxidativa, marcadores inflamatorios y daño mitocondrial. Existe cada vez más evidencia sobre el protagonismo de la vitamina D en la regulación del sistema renina-angiotensina-aldosterona (RAAS), con posibles implicancias cardiovasculares así como para el sistema inmunológico. Valores de vitamina D inferiores a 25 ng/mL se relacionan con un aumento de tono vascular mediado por la contracción del músculo liso, ya sea a través de efectos directos sobre las células vasculares del músculo liso, sobrerregulación del RAAS, y/o a través de la modulación del metabolismo del calcio con hiperparatiroidismo secundario; lo cual predispone en los pacientes a desarrollar hipertrofia del ventrículo izquierdo y de la pared vascular originando HTA. En este trabajo se realizó una revisión de los principales mecanismos involucrados en el desarrollo de la HTA asociados al déficit de vitamina D. Entre ellos se destaca el vínculo que se establece entre los niveles de fosfato inorgánico extramitocondrial, sus principales hormonas reguladoras—como la vitamina D—, el aparato cardiovascular, las especies reactivas del oxígeno y el metabolismo mitocondrial. El papel del receptor de vitamina D a nivel mitocondrial y la regulación de la cadena respiratoria influirían en el remodelamiento arterial, ya que su activación reduciría el daño oxidativo y preservaría la vida celular. No obstante, existen aspectos aún no comprendidos sobre la intrincada red de señalización que resultan simples en ensayos experimentales, pero complejas en los estudios clínicos y donde la concreción de nuevos estudios como el VITAL podría clarificar, y así apoyar o refutar los posibles beneficios de la Vitamina D en la enfermedad cardiovascular hipertensiva.
Classically, Vitamin D (VitD) plays a major role in controlling bone metabolism. It is obtained from diet, exposure to the sun and dietary supplements. However, in recent decades, it has gained importance as a modulating factor in the response of cells by inducing their proliferation in many tissues, including at the level of the cardiovascular system; and closely related, a significant VitD deficiency affecting almost 50% of the population worldwide has been shown.1 This hypovitaminosis D pandemic—in principle—can be mainly attributed to factors that reduce exposure to sunlight, which is necessary for the formation of dermal precursors of VitD by the action of ultraviolet B radiation (UVB). Here, lifestyle and environmental factors such as reduced outdoor activities and environmental pollution are highlighted. In fact, while humans can obtain VitD from the diet, the most important source is exposure to sunlight, which varies with the season and latitude. To be biologically active, VitD must undergo two sequential hydroxylations—hepato-renal—to eventually become the active metabolite 1,25-dihydroxycholecalciferol (1,25(OH)2D3). Race is another relevant aspect to consider; people with black skin absorb more UVB rays through their skin’s melanin, therefore, they require more exposure to produce the same amounts of VitD.2 It is noteworthy that all conditions associated with low levels of VitD (poor UVB induction), whether due to high latitude, industrialisation, or dark skin, have also been associated with increased blood pressure.2 Furthermore, VitD deficiency has been associated with myocardial infarction, stroke and other cardiovascular diseases, including atherosclerosis and endothelial dysfunction.3 A close relationship has been observed recently between osteoporosis and cardiovascular disease, which appears to be associated with VitD deficiency. Of particular interest, numerous cross-sectional and prospective studies show that levels below 25 ng/mL are associated with both diseases.4 In this regard, there is increasing evidence on VitD and its role in the regulation of the renin-angiotensin-aldosterone system (RAAS), with possible cardiovascular implications as well as at the level of the immune system (Fig. 1).

The word cloud as a summary of the topic highlights, according to the size and distribution of the words, the relevant implications that are established between hypertensive cardiovascular disease and vitamin D levels in the context of basic studies and their opposite number, clinical studies.
As mentioned above, Vit D deficiency has been associated with increased vascular tone mediated by smooth muscle contraction. In this regard, it was observed that children with primary hypertension had, in 71% of cases, VitD levels lower than 20 ng/mL.5 Here, multiple mechanisms would be overlapped by vascular smooth muscle cell alterations, elevated expression of RAAS mediators, calcium metabolism disturbances with the development of hyperparathyroidism in patients complicated with left ventricle hypertrophy, vascular remodelling and arterial hypertension (AHT).6
In murine models with VitD receptor (VDR) knockout, a higher incidence of hypertension, left ventricular hypertrophy and atherosclerosis was observed,7 while in animals with AHT (SHR), the use of VitD or analogues reduced left ventricular hypertrophy in an equivalent manner to losartan.8 On the other hand, in normal mice, VitD deficiency stimulated renin expression, while its injection reduced renin synthesis. In addition, studies in cell cultures showed that this relationship was due to an inhibitory process of VitD on renin gene transcription, through a VDR-dependent mechanism. This was confirmed because the mice lacking the VDR gene developed hyperreninaemia, resulting in high angiotensin II (Ang II) production, which inexorably led to hypertension, cardiac hypertrophy and increased water intake.8
VDR knockout in mouse endothelial cells showed an altered response to endothelium-dependent relaxation and exaggerated response to Ang II infusion. This anomaly would correspond—among other factors—to nitric oxide (NO) signalling pathway impairment. The cardiovascular response would be mediated by the effect of VitD on the endothelium, and its deficiency would lead to AHT.9 It was also observed that diabetic and VDR knockout (VDR-KO) mice developed more severe nephropathy than wild mice, suggesting that VitD would protect against kidney damage by regulating the RAAS.10 Closely related and with translational implications, a meta-analysis by Derakhshanian et al. suggested that VitD would play a nephroprotective role, although clinical trials have not shown significant benefit from its use, or from analogues such as paricalcitol.11 Of interest, with regard to paricalcitol and calcium metabolism, the effect of VitD on renin is independent of calcium metabolism.12 More specifically, Kong et al. worked with a model of transgenic mice that overexpress human VDR to evaluate renin-producing cells and demonstrated that suppression of renin expression by VitD in vivo is independent of parathyroid hormone and of calcium.13
Inappropriate activation of the RAAS has been reported in VDR-KO and 1α-hydroxylase-KO mice14,15 these mice developed AHT and myocardial hypertrophy, which continued present even after calcium homeostasis was normalised; however, RAAS blockade with angiotensin- converting enzyme inhibitors normalised blood pressure and cardiac disturbances. On the other hand, increased activation of the RAAS, AHT and myocardial disturbances could be successfully treated with VitD in 1α-hydroxylase knockout mice. Renin activation with low levels of VitD has been confirmed in patient cohort studies. These data suggest that plasma levels of VitD could result in positive RAAS regulation.16,17 The molecular effects of VitD on the RAAS are now clearer due to the landmark discovery that when VitD binds to the cAMP (CREB) response element binding protein, it prevents the formation of a CRE-CREB-CBP/p300 complex and this complex suppresses renin gene expression. As a result, renin expression decreases because CREB can no longer bind to the cAMP response element, and therefore, is not able to activate transcription in the renin gene promotor region.18
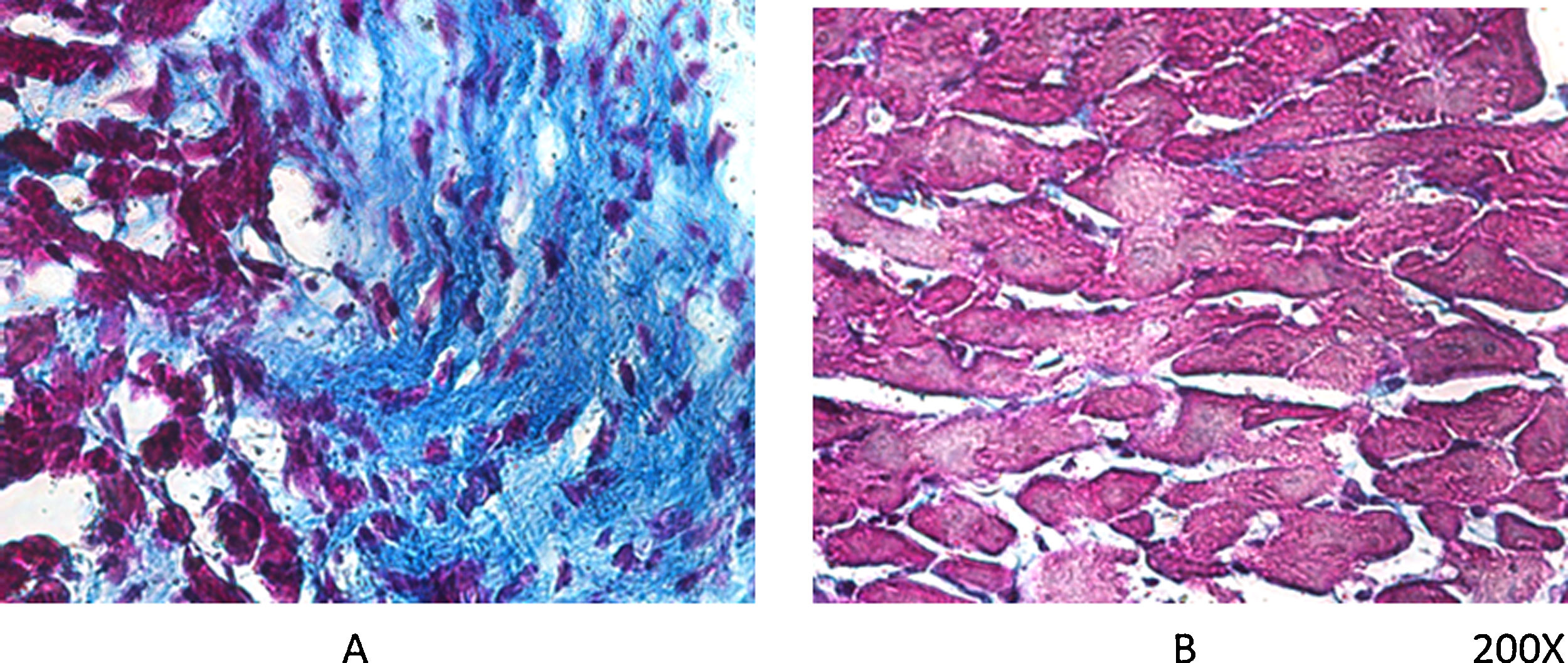
As mentioned earlier, VDR-KO mice show overexpression of myocardial renin and marked hypertrophy of their cardiomyocytes. Simpson et al. also showed that VitD-deficient animals had increased incidence of AHT, presented left ventricular hypertrophy, and developed atherosclerosis.19 In the PRIMO trial, which included 227 patients with stage 3–4 chronic kidney disease who were randomised to receive paricalcitol or placebo, change in left ventricular mass index after 12 months did not differ between the two groups.20 However, a post hoc study of the PRIMO trial showed that 48 weeks of therapy with paricalcitol significantly reduced left atrial volume, which would be associated with reduced mortality.21 In this regard, our laboratory observed a reduction myocardial VDR expression in rats with obstructive nephropathy, which could be related to myocardial remodelling associated with increased arrhythmogenesis. However, the use of paricalcitol protected against these changes by restoring myocardial VDR levels, reducing the fibrotic process and prolonging action potentials22 (Fig. 2).

Regarding the molecular mechanisms involved in cardiovascular disease and the decoupling of the RAAS, Klotho plays a leading role. Its cross-talk exerts a significant influence on the development of cardiovascular disease in animal models of AHT, diabetes and chronic kidney disease. Klotho factor is a transmembrane protein involved in the regulation of calcium and phosphate metabolism. It is produced in various tissues and is expressed in soluble form or anchored to the plasma membrane. It was initially discovered as a protein with an anti-aging function. Three types are known: α, β, and γ. Klotho exerts its action by acting as a cofactor of a crucial protein of the fibroblast growth factor family called fibroblast growth factor 23 (FGF23). FGF23 is a phosphatonin encoded at the 12p13 locus that promotes phosphate excretion in the proximal contoured tubule by inhibiting the NaPi co-transporter located in the luminal membrane. Its action is triggered after binding to the FGFR1 receptor only if it is associated with the Klotho protein. In addition, cross-talk between FGF23 and Klotho not only regulates phosphataemia, but also decreases renal production of VitD by inhibiting the 1α-hydroxylase enzyme. VitD, in turn, is able to increase the expression of FGF23 and Klotho by modulating the transcription of their genes. The potential regulation of klotho mRNA by VitD was deduced from studies reported by Tsujikawa et al.,23 who described the effect of pharmacological and dietary manipulations of VitD in mice on the expression of the klotho messenger of 5.2 kb.24 Disturbances that condition reduction in the expression and activity of FGF23 and Klotho are associated with acceleration of cell aging processes, vascular and soft tissue calcification and increased mortality, while normalisation of serum phosphate levels reverses these pathological changes.25–27 Likewise, low levels of Klotho cofactor have been reported to be associated with cardiovascular disease, ventricular hypertrophy and increased mortality.28 Genetic deficiency of Klotho factor leads to cardiac fibrosis and the development of left ventricular hypertrophy. Left ventricular dysfunction is detected before hypertrophy and fibrotic changes in the myocardium occur. Klotho is able to exert a counter-regulation on the pro-fibrotic action of TGFβ and Ang II. The molecular pathways involved in the processes of hypertrophy and fibrosis are the same that lead to these disturbances at the renal level.29 Of central interest to this review is that serum phosphate levels also significantly impact mitochondrial metabolism. Inorganic phosphate regulates oxidative phosphorylation at various levels, which is evidenced by the fact that the increase of extramitochondrial phosphate in porcine cardiac and skeletal muscle stimulates oxygen consumption in the mitochondria, increases NADPH levels and regulates transmembrane potential.30 in addition, the increased influx of reduction equivalents to the cytochrome III complex triggered by higher phosphate levels results in increased production of reactive oxygen species (ROS) and as a consequence increases mitochondrial oxidative stress.31 This is a relevant axis and indicates the link established between levels of extramitochondrial inorganic phosphate, its main regulatory hormones—VitD, parathyroid hormone (PTH), FGF23 and Klotho cofactor-, the cardiovascular system, ROS and mitochondrial metabolism.
Ang II, on the other hand, by binding to the AT1 receptor, leads to an increase in ROS production by the activation of the NADPH oxidase enzyme.31,32 It also stimulates the production of NO, which ultimately combines with ROS, particularly with the O2− superoxide anion to form peroxynitrites.33 This leads to further oxidative damage and impairs the availability of NO. In pathophysiological situations with RAAS hyperactivation, such as during hypertensive disease, the exacerbated production of Ang II assumes a key role in the generation of ROS and contributes to cell and tissue damage.
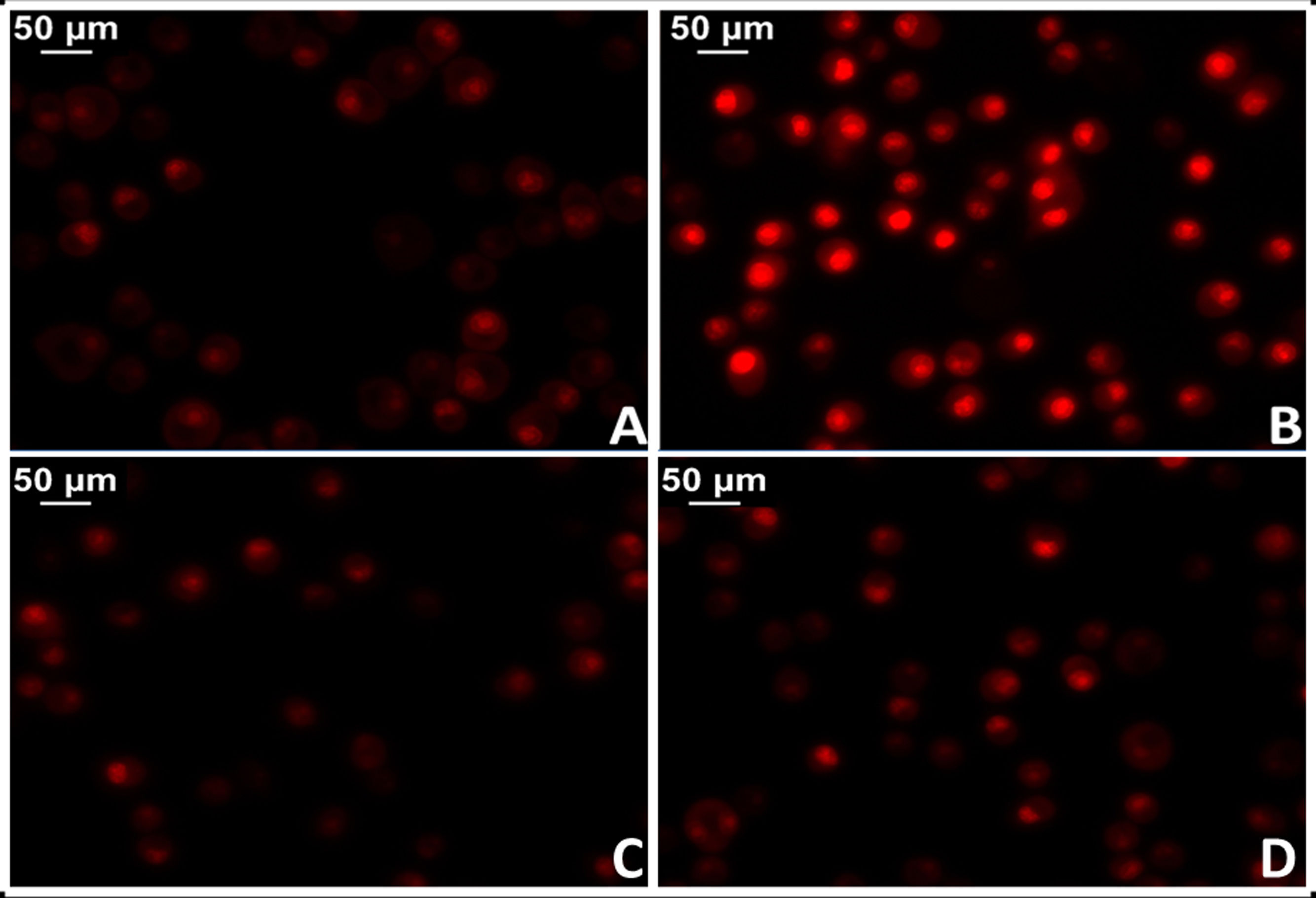
The administration of Ang II causes increased protein oxidation, increased mitochondrial DNA content and genetic deletions in the mitochondria of the heart muscle of mice, while these harmful effects are not shown in mouse models with genes that overexpress mitochondrial catalase (MCAT). Similarly, cardiac hypertrophy was less in these mice.34 In this regard, there is strong evidence that shows activation of mitochondrial NADPH oxidase by the action of Ang II and increased production of mitochondrial ROS.35 In agreement, blocking Ang II decreases ROS production and improves mitochondrial function. More specifically, the implementation of losartan in spontaneously hypertensive (SHR) rats succeeded in preventing the production of H2O2, mitochondrial superoxide dismutase activity and cytochrome oxidase.36,37 In this sense, preliminary results from our laboratory in myocardial tissue of losartan-treated SHR rats showed a significant reduction in mitochondrial damage with reduction of superoxide anion levels (Fig. 3).
 Myocardial tissue from normotensive (WKY) rats. B) Myocardial tissue from spontaneously hypertensive rats (SHR) C) Myocardial tissue from normotensive rats treated with losartan at a rate of 40 mg/kg/day in their drinking water (WKY + Los) D) Myocardial tissue from spontaneously hypertensive rats treated with losartan at a rate of 40 mg/kg/day in their drinking water.")
Test for oxidative cell stress in rat myocardial tissue using the fluorescent dye dihydroethidium technique to evaluate superoxide anion production. A) Myocardial tissue from normotensive (WKY) rats. B) Myocardial tissue from spontaneously hypertensive rats (SHR) C) Myocardial tissue from normotensive rats treated with losartan at a rate of 40 mg/kg/day in their drinking water (WKY + Los) D) Myocardial tissue from spontaneously hypertensive rats treated with losartan at a rate of 40 mg/kg/day in their drinking water.
In mitochondrial metabolism it has been described that VDR inhibits the respiratory chain. VDR silencing improved the respiratory activity of cells. VDR induction reduces the transcription of proteins from the respiratory chain and the mitochondrial membrane potential, diverting the metabolism and optimising the consumption of energy, while the loss or reduction in VDR expression results in significant cell growth inhibition, with accumulation in the G0 and G1 phases of the cycle and reduction of the M phase. The effects of VDR silencing on proliferation were confirmed in several human cancer cell lines. A distinctive feature of tumour cells is impaired metabolism that supports rapid cell proliferation, with the mitochondria being responsible for many impaired metabolic processes that increase cell proliferation.38 In parallel, VDR is involved in mitochondrial metabolic modulation in fatty tissue. Specifically, at the level of fat tissue the mitochondrial action of VDR is exerted on the metabolism of fatty acids. In studies conducted in transgenic rats, VDR overexpression led to decreased expression of decoupled proteins, decreased beta-oxidation and lipolysis. This metabolic decoupling was accompanied by a decrease in coding genes for key enzymes in these signalling pathways, such as hexokinase, carnitine palmitoyl transferase and lipase. In these mice, an increase in adipose tissue gain was observed, accompanied by low levels of leptin and high levels of cholesterol. In contrast, in VDR-KO mice an increase in the expression of decoupled proteins in brown adipose tissue was observed, together with an increase in the expression of these proteins in VitD-treated brown adipocytes, suggesting an inhibitory action of VitD on the decoupled proteins and a decrease in the use of lipids, with a redirection of acetyl-CoA towards biosynthetic pathways.39
In relation to previous concepts, one of the genomic effects of VitD is to decrease the production of prorenin by inhibiting its transcription, leading to less RAAS activation. Ang II reduces Klotho expression, and this causes the FGF23/Klotho ratio to increase, which conditions a reduction in VitD levels and also negatively impacts the regulation of the RAAS, reinforcing its activation.40 Therefore, it can be concluded that VitD intervenes decisively in the control of the oxidation-reduction imbalance induced by hyperactivation of the RAAS.
Another molecular mediator that plays a leading role in relation to VitD are the so-called sirtuins. In this regard, VitD has been shown to reduce oxidative stress by increasing the expression of sirtuin 1 (SIRT1) and AMPK.41 The sirtuin family comprises a set of 7 proteins involved in the modulation of processes linked to the development of diseases associated with aging and exerts multiple functions in metabolism and cell proliferation.26 They are basically enzymes with deacetylase activity that depend on intracellular levels of NAD +. Sirtuins and VitD interrelate in a dual manner because they reciprocally stimulate their expression in a positive feedback cycle.42 More specifically, sirtuin 3 (SIRT3) modulates the functioning of the urea cycle and the oxidation of fatty acids in the mitochondrial matrix. It also exerts a protective influence on the mitochondrion by virtue of its ability to create a reducing environment through increased activity of the glutathione reductase enzyme, which prevents the harmful effects of oxidative stress. Thus, the ratio of available reduced glutathione to oxidised glutathione derived from the reduction of harmful species is increased in favour of the former.43 In this regard, it has been shown that RAAS hyperactivation with Ang II elevation—such as that present in cardiovascular diseases—alters the functions of SIRT3 inhibiting its expression.44 On the other hand, SIRT1 increases the expression of angiotensin converting enzyme type 2 (ACE2). In this regard, the anti-inflammatory, anti-proliferative and vasodilatory effects of excision products of ACE2 on the physiopathological profile of cardiovascular disease are widely known. Differential processing of angiotensin precursors by the pathway governed by ACE2 produces angiotensin 1–7 (Ang 1–7) capable of activating a Mas receptor (MasR) that executes its effects on target organs. Of interest, very recently it has been demonstrated in hypertensive rats that the use of a specific MasR antagonist nullified the antioxidant and immunomodulatory actions of VitD. These findings indicate the involvement of the ACE2I/Ang 1–7/MasR pathway in the protective mechanisms of VitD.45
All these data continue to provide evidence of the non-genomic effects of VitD, in addition to the classic mechanism where VDR acts as a transcription factor that migrates to the nucleus through the pores of the nuclear membrane. However, a new mechanism of action of VitD has recently been suggested related to the intramitochondrial location of its VDR39 receptors.39
In this regard, the presence of the VitD receptor in human platelets has been demonstrated in their mitochondria using anti-VDR antibodies. Platelets are an ideal sample for the study of the extranuclear location of VDR since they lack a nucleus.46 Progress was made in this regard, and in experimental models with RAAS induction and AT1 receptor overexpression it could be shown that the use of paricalcitol caused reduction of AT1 expression and intramitochondrial VDR induction, suggesting a direct cytoprotective link between paricalcitol and the AT1 receptor mediated by activation of mitochondrial VDR.47 Subsequently, our laboratory proposed that this decrease in VDR- induced AT1 could be a consequence of cell protection mediated by heat shock protein 70 (Hsp70).48 In fact, we observed in the mitochondria a significant association in the expression of AT1, VDR and Hsp70. Prior to our findings, it was determined that Hsp70 cross-talks with VDR and plays a role in controlling its intracellular concentration.49 It has also been demonstrated in rat kidney that VitD is an inducer of Hsp70.50 Closely related, it was possible to establish, in a model of cardiovascular involvement with kidney failure, significant prevention of kidney damage and myocardial remodelling associated with reduction in oxidative stress, fibrosis and apoptosis with the reduction of AT1 and increases in Hsp70 and VDR.51
In this regard, it is worth mentioning that—in rat kidney embryonic cells, from primary Wilms' tumour samples, as well as in cells cultured with inducible expression of Wilms' tumour protein 1 (WT-1)—an association between Hsp70 and WT-1 has been previously demonstrated.52 This protein is required for nephrogenic and cardiogenic processes. Of interest, Hsp70 has been described as an important cofactor for WT-1 function, and a potential role for this chaperone during the nephrogenic process was suggested. In addition, VDR messenger and protein have been identified in embryonic kidneys, which suggests a role for the VitD system in kidney development. The temporary expression patterns of WT-1 and VDR genes correlate closely in the kidney during development. Promoters of human and mouse VDR genes have also been shown to contain several consensus sites of binding to WT-1.53,54
Clinical and epidemiological studiesThe prevalence of AHT in 2000 was 24.6% worldwide and based on projections it is estimated to reach 29.2% of the population in 2025.55 AHT is a prevalent disease that mainly affects the heart, kidneys and brain. Better management and new treatments could reduce damage to these target organs. In terms of mechanisms and possible new approaches, a meta-analysis prior to 2015 that included 8 prospective studies reported that VitD levels were inversely associated with the incidence of AHT (RR: .70; 95% CI: .58 to .86).56 This was observed in clinical studies conducted over the past two decades that have highlighted an inverse association between plasma levels of 1.25(OH)2D3, blood pressure and/or plasma renin activity, both in normotensive males and in patients with essential hypertension.16,57
Some more recent meta-analyses observe studies with positive results in the treatment of blood pressure. Taking into account the cut-off point of 20 ng/mL (or 50 nmol/L) as a VitD deficiency, favourable results were observed with VitD supplementation in obese individuals (BMI greater than 30 kg/m2) and hypertensives over 50 years of age with VitD-deficiency levels below 20 ng/mL (50 nmol/L).58 This is associated with decreased VitD absorption in the elderly.
In an adult population sample, Sabanayagam et al. found that lower levels of serum VitD were associated with prehypertension, with a relative risk of 1.48 (CI 1.16–1.90).59 VitD deficiency as an environmental risk factor favours increased vascular tone, which may not play a major role in regulating normal blood pressure homeostasis, but serves as a trigger contributing to the development of AHT in vulnerable middle-aged individuals.60
Experimental studies in mice have indicated that VitD supplementation significantly reduces renin synthesis and blood pressure. It is possible and conceivable that similar mechanisms of action could be found in humans. However, the expected results have not yet been obtained. Nonetheless, in line with the abovementioned data, some studies have shown a reduction in blood pressure in patients with primary hypertension who received VitD supplementation,61 and a reduction in blood pressure, plasma renin and Ang II levels in patients with secondary hyperparathyroidism.62,63 Treatment with VitD achieved a 50% reduction in renin mRNA compared to the control group. These findings coupled with the experimental results suggest the importance of VitD as an effective suppressor of renin synthesis.64 In addition, other trials have demonstrated the greater effectiveness of paracalcitol treatment compared to Vit D supplementation. Among its observed benefits: reduced renin levels, decrease of interleukins 1 and 6, tumour necrosis factor and proteinuria levels demonstrating a nephroprotective effect.65
The role of the inflammatory pathway and VitD-mediated signalling in cardiovascular disease is also exposed. Of particular interest, VDR signalling in leukocytes and macrophages places an important role in the development of atherosclerosis and AHT. At least part of the anti-atherosclerotic mechanism is to block the activation of the local RAAS in macrophages and within the atherosclerotic lesion.66
In the context of the development of atherosclerosis, the involvement of the aorta and its wall determines the onset of aortic aneurysms, and closely related to the central theme of this review, aortic aneurysms are strongly associated with AHT and the RAAS, as shown by previous studies. Specifically, AHT determines the occurrence of diseases of the artery wall, such as atheroma, dissections and aneurysms. The latter are important in relation to their site and severity. Aortic aneurysm is characterised by progressive degeneration of the structure of the artery wall by chronic inflammation, atherosclerosis and inherent factors that determine changes with remodelling of the wall and that lead to dilation.67 Dilation as it progresses becomes irreversible and rupture can be clinically fatal. Thus, some patients survive the picture while others present as sudden death. Of particular interest, the disturbances caused have also been related to VitD deficiency and progression of AHT in its natural evolution. This was evidenced in a follow-up of patients with aneurysm versus controls (reviews and meta-analyses).68–70
Regarding the possible molecular mechanisms responsible, there is evidence to suggest a close relationship between VitD and regulation of the RAAS as mentioned earlier. More specifically, mice were used as a model of atherosclerosis due to apolipoprotein E deficiency (ApoE−/−) and the formation of abdominal aortic aneurysms was also induced in them by the administration of Ang II. As a relevant result we found that oral treatment with calcitriol reduced the formation of aneurysms with a marked increase in the interaction of VDR-RXR-α (retinoid X receptor α) in the aortas of calcitriol-treated mice. In addition, they exhibited reduced infiltration of macrophages, matrix metalloproteinase (MMP) and expression of chemokines in the adrenal aortic walls, apparently as a consequence of the activation of VDR-RXR-α interactions.71 This study provided strong evidence that chronic treatment with calcitriol would reduce RAAS excitation- induced aneurysm formation because Ang II is one of its main exponents. These effects would be associated with reduced inflammatory response and regulation of extracellular matrix homeostasis through VDR-RXR-α heterodimerisation. In light of these results, activation of VDR could represent a promising therapeutic target for the treatment of aortic aneurysm.71
In parallel, the effects exerted by the activation of VDR play a critical role in modulating the signals involved in atherosclerotic processes. In incipient atherosclerotic lesions, differentiation of T lymphocytes towards the Th1 phenotype drives macrophage activation through the production of IFNγ. The phagocytosis of modified LDL particles and their accumulation in the subintimal region of the arterial wall ultimately forms fat striation precursor to established atherosclerotic lesions. VitD is able to interfere with this pathological process through regulation of the differentiation of cooperating T-lymphocytes. In this regard, it has been observed that VitD analogues induce the change to a Th2 phenotype in T lymphocytes in the presence of IL 4 action. In turn, VitD analogues possess an inhibitory effect on IFNγ production, leading to decreased Th1 lymphocyte differentiation and decreased phagocyte cell recruitment.72 Increased VDR action also suppresses the entry of oxidised LDL particles towards the arterial intima and depresses foam cell formation.73 As a fundamental aspect, previously cited, the action of VitD in the anti-atherosclerotic process seems to also depend on the reduced activation of the local RAAS in atheromatous plaque. This is because Ang II, through the occupation of AT1 receptors, promotes the expression of proinflammatory genes and increases mitochondrial oxidative stress as a result of chronic activation of the NFκB pathway and the NADPH oxidase enzyme.74
In parallel, atherosclerosis as a chronic disease is associated with cardiovascular dysfunction including myocardial infarction, unstable angina, sudden cardiac death, stroke, aortic aneurysms and peripheral thrombosis. Atherosclerosis is predicted to be a leading cause of death worldwide by 2020 due to clear endothelial damage by oxidative stress associated with cardiovascular risk that include diabetes and AHT, among the most prominent.75
In this regard, Krause et al.76 conducted a study in which participants were exposed to UVB radiation on a sunbed 3 times per week for 3 months. The subjects had a 180% increase in their VitD levels and a reduction of 6 mmHg in both systolic and diastolic blood pressure. A small randomised, placebo-controlled study in patients with type 2 diabetes (DM2) and low baseline levels of VitD subsequently showed that a single dose of 100,000 IU VitD reduced systolic blood pressure by 14 mmHg, and the same dose significantly improved endothelial function as measured by forearm blood flow.77 In the NHANES3 study, mean systolic blood pressure was approximately 3 mmHg lower in individuals in the highest quintile of serum VitD compared to those in the lowest quintile.78 However, although several epidemiological studies have shown that there may be an association between hypertension and low VitD levels, the results do not yet support the use of VitD or its analogues as individual treatment for patients with hypertension or as a population-level intervention to reduce blood pressure.79
Scragg et al.78 published their findings on the relationship between VitD levels and blood pressure. The authors studied the data from NHANES3 and found a significant inverse relationship between VitD levels and blood pressure levels, which was evident even after adjusting for variables such as age, sex, ethnicity, and physical activity. Judd et al.80 also analysed data from NHANES3 and showed a statistically significant inverse relationship between circulating VitD levels and systolic blood pressure. Martins et al.,81 also from NHANES3 data, found that a low level of VitD was associated with an increased risk of hypertension. These results would be possible justifications for Ludwigshafen Risk and Cardiovascular Health (LURIC) studies. Here the aim was to document a possible association between different types of circulating VitD and RAAS in a large patient cohort (n = 3316) referred for coronary angiography. After measuring 25(OH)D3, 1,25(OH)2D3, plasma renin and Ang II concentration, they were able to demonstrate for the first time in humans, an independent association between them.82 However, the association in a reduction in AHT and VitD supplementation had also been evaluated previously. Thus, in a meta-analysis of studies between 1966 and 2014 it was concluded that blood pressure did not drop with this treatment, and did not decrease significantly, and therefore its use as an antihypertensive would not be recommended.83
Therefore, given the extent of controversy and in order to investigate in detail a possible relationship between blood pressure and VitD levels, the VITAL study is currently under development. Its primary objectives are to determine whether supplementation with VitD and fish oil lowers 24-h ambulatory blood pressure compared to placebo in a sub-cohort of 1000 participants. The study also seeks to evaluate whether VitD and fish oil supplementation reduces the risk of incident hypertension compared to placebo among all randomised VITAL participants without baseline hypertension, and whether VitD and fish oil supplementation favourably changes hypertension-related biomarkers compared to placebo. A representative sub-cohort of 1000 non-hypertensive participants in the VITAL study from selected major metropolitan areas in the United States will also be invited to participate in study visits in their home at the start of the study and after 2 years of follow-up. During these visits, participants will be asked to use monitors to record 24-h ambulatory blood pressure measurements, provide fasting blood, spot urine samples, and other clinical measurements. The results are expected after the study closes in November 2020. Undertaking these studies will provide more relevant evidence to support or refute the possible preventive roles of VitD and omega-3 fatty acids in blood pressure and the development of hypertension.84
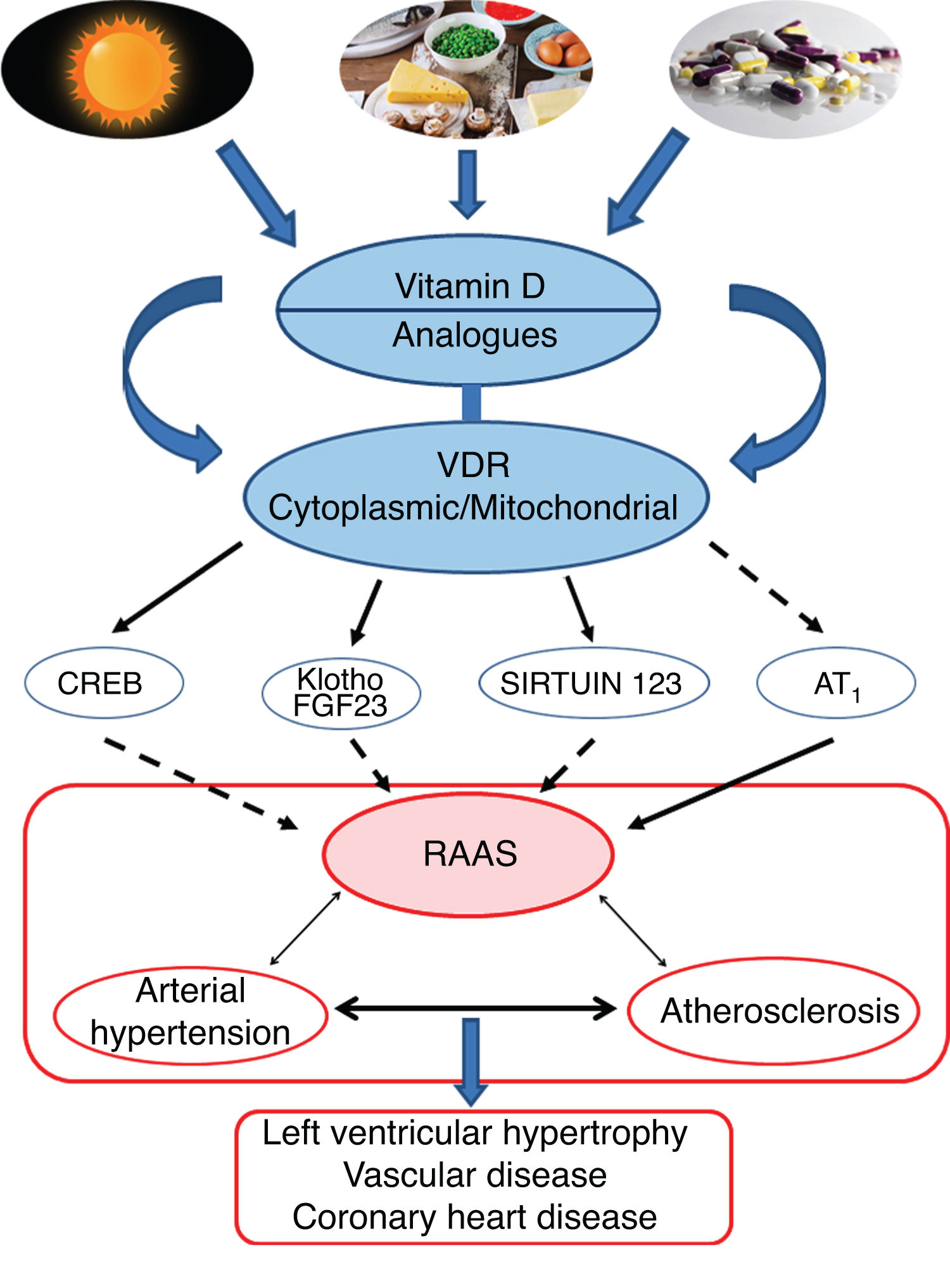
Conclusions and perspectivesThe leading role of VitD and its protective effect against AHT would essentially be supported by its effects in negatively regulating the RAAS pathway. More specifically, the VDR would modulate the respiratory chain at the mitochondrial level, which would influence cardiovascular remodelling. Activation of the VDRs would reduce oxidative damage and preserve cell and tissue viability. Animal studies have clearly shown that VitD deficiency is associated with AHT, atherosclerosis and cardiac hypertrophy (Fig. 4).

Graphic/schematic summary of the main signalling pathways involved in the modulation of vitamin D—and its receptors, both cytoplasmic and mitochondrial—on the RAAS and their impact on the development of cardiovascular disease and its main exponents represented here as arterial hypertension, atherosclerosis and coronary disease. The solid arrows indicate induction/stimulation, while the dashed arrows indicate inhibition.
In contrast, clinical trials show contradictory and non-categorical results, which can in part be attributed to their design. Studies establishing the role of VitD in the framework of current treatment for AHT are therefore desirable. It is important to establish the actions and results of treating patients with VitD in the different stages of hypertensive disease, in order to apply the benefits proven in basic research studies.
VDRs are present in vascular smooth muscle, endothelium, cardiac muscle cells, and even within the main organelle responsible—when dysfunctional—for cardiovascular hypertensive disease, the mitochondria. Observational studies have shown a relationship between low levels of VitD, blood pressure and even calcification of the coronary arteries. Low VitD levels are associated with increased oxidative load, inflammatory markers, and mitochondrial damage. Some animal and human studies have indicated that VitD may play a key role in glucose homeostasis and the development of diabetes.
Finally, undertaking studies such as VITAL may clarify as yet not understood aspects of the intricate signalling network, which are simple in experimental trials but complex in clinical studies, in order to support or refute the potential benefits of VitD and hypertensive cardiovascular disease.
FundingThis work received financial support from the Research and Technology Council of the University of Cuyo (SECyT), Mendoza, Argentina, and from ANPCyT FONCyT, both awarded to Walter Manucha. Grant No. PICT 2016-4541.
AuthorshipAll the authors contributed equally to the conception and design of the review, with substantial input on data, analysis and interpretation of content, writing and critical review of the article for its intellectual content.
Conflict of interestsThe authors have no conflict of interest to declare.
Please cite this article as: Sanz R, Mazzei L, Santino N, Ingrasia M, Manucha W. La interacción vitamina D-mitocondria podría modular el camino de señalización involucrado en el desarrollo de la hipertensión: una visión integrativa translacional. Clin Investig Arterioscler. 2020;32:144–155.