




El déficit de vitamina D es una pandemia a nivel mundial y trae como consecuencia osteoporosis, hipertensión arterial (HTA) y otras enfermedades cardiovasculares. A nivel celular produce mayor carga oxidativa, marcadores inflamatorios y daño mitocondrial. Existe cada vez más evidencia sobre el protagonismo de la vitamina D en la regulación del sistema renina-angiotensina-aldosterona (RAAS), con posibles implicancias cardiovasculares así como para el sistema inmunológico. Valores de vitamina D inferiores a 25ng/mL se relacionan con un aumento de tono vascular mediado por la contracción del músculo liso, ya sea a través de efectos directos sobre las células vasculares del músculo liso, sobrerregulación del RAAS, y/o a través de la modulación del metabolismo del calcio con hiperparatiroidismo secundario; lo cual predispone en los pacientes a desarrollar hipertrofia del ventrículo izquierdo y de la pared vascular originando HTA. En este trabajo se realizó una revisión de los principales mecanismos involucrados en el desarrollo de la HTA asociados al déficit de vitamina D. Entre ellos se destaca el vínculo que se establece entre los niveles de fosfato inorgánico extramitocondrial, sus principales hormonas reguladoras —como la vitamina D—, el aparato cardiovascular, las especies reactivas del oxígeno y el metabolismo mitocondrial. El papel del receptor de vitamina D a nivel mitocondrial y la regulación de la cadena respiratoria influirían en el remodelamiento arterial, ya que su activación reduciría el daño oxidativo y preservaría la vida celular. No obstante, existen aspectos aún no comprendidos sobre la intrincada red de señalización que resultan simples en ensayos experimentales, pero complejos en los estudios clínicos. En este sentido, la concreción de nuevos estudios como el VITAL podría clarificar, y así apoyar o refutar, los posibles beneficios de la vitamina D en la enfermedad cardiovascular hipertensiva.
Vitamin D deficiency is a worldwide pandemic and results in osteoporosis, hypertension, and other cardiovascular diseases. At the cellular level, it produces significant oxidative stress, inflammatory markers, and mitochondrial damage. There is increasing evidence about the role of vitamin D in the regulation of the renin-angiotensin-aldosterone system (RAAS). Moreover, there is evidence of involvement in cardiovascular complications, as well as in the immune system disorders. Vitamin D values below 25ng/mL are related to an increase in vascular tone mediated by smooth muscle contraction. Furthermore, it can produce direct effects on vascular smooth muscle cells, RAAS over-regulation, modulation of calcium metabolism, and secondary hyperparathyroidism. All this predisposes patients to develop hypertrophy of the left ventricle and vascular wall, causing hypertension. In this work, a review is presented of the main mechanisms involved in the development of hypertension due to vitamin D deficiency. Among them are the link established between the levels of extra-mitochondrial inorganic phosphate, its main regulatory hormones —such as vitamin D—, the cardiovascular system, reactive oxygen species, and mitochondrial metabolism. The role of the mitochondrial vitamin D receptor and the regulation of the respiratory chain could influence arterial remodelling since its activation would reduce oxidative damage and preserve cell life. However, there are aspects not yet understood about the intricate signalling network that appeared simple in experimental trials, but complex in clinical studies. In this way, the completion of new studies as VITAL, could clarify, and thus support or refute the possible benefits of vitamin D in hypertensive cardiovascular disease.
Clásicamente la vitamina D (VitD) resulta protagónica en el control del metabolismo óseo. Se obtiene de la dieta, exposición solar y suplementos dietarios. No obstante, en las últimas décadas, ha cobrado importancia como factor modulador de la respuesta de las células al inducir su proliferación en muchos tejidos, incluido a nivel del aparato cardiovascular; y en estrecha relación, se ha demostrado que existiría una significativa insuficiencia de VitD que afectaría a casi el 50% de la población en todo el mundo1. Esta pandemia de hipovitaminosis D —en principio— puede atribuirse principalmente a factores que reducen la exposición a la luz solar, necesaria para la formación de precursores dérmicos de VitD por acción de la radiación ultravioleta B (UVB). Aquí se destacan el estilo de vida y el ambiente, como la reducción de actividades al aire libre y la polución ambiental. De hecho, si bien el hombre puede obtener VitD de la dieta, la fuente más importante es la exposición al sol, que varía según la estación del año y la latitud. Para ser biológicamente activa, la VitD debe sufrir dos hidroxilaciones secuenciales —hepato-renal— para convertirse finalmente en el metabolito activo 1,25-dihidroxicolecalciferol (1,25(OH)2D3). Otro aspecto relevante a considerar en el hombre es en relación con las distintas razas; los individuos de piel negra absorben más rayos UVB a través de la melanina de su piel, por lo tanto, requieren más exposición para producir las mismas cantidades de VitD2. Para destacar, resulta notorio que todas las condiciones asociadas a bajos niveles de VitD (pobre inducción UVB), sea por elevada latitud, industrialización, así como por piel oscura, han sido asociadas también con un aumento de los valores de presión arterial2. Más aún, la deficiencia de VitD ha sido relacionada con infarto de miocardio, accidente cerebrovascular y otras enfermedades cardiovasculares, incluyendo aterosclerosis y disfunción endotelial3. Recientemente se ha observado una relación estrecha entre la osteoporosis y la enfermedad cardiovascular, lo cual parece estar asociado al déficit de VitD. De especial interés, numerosos estudios transversales y prospectivos ponen en evidencia que valores inferiores a 25ng/mL se asociarían con ambas patologías4. En este sentido, existe cada vez más evidencia sobre VitD y su protagonismo en la regulación del sistema renina-angiotensina-aldosterona (RAAS), con posibles implicancias cardiovasculares así como también a nivel del sistema inmunológico (fig. 1).

La nube de palabras como resumen del tema destaca, según el tamaño y la distribución de las palabras, las implicancias relevantes que se establecen entre la enfermedad cardiovascular hipertensiva y los niveles de vitamina D en el contexto de los estudios básicos y su contracara, los estudios clínicos.
Como se ha mencionado previamente, el déficit de VitD ha sido relacionado con un aumento del tono vascular mediado por la contracción del músculo liso. Al respecto, se observó que niños con hipertensión primaria tenían, en un 71% de los casos, valores de VitD inferiores a 20ng/mL5. Aquí, múltiples mecanismos estarían solapados por alteraciones sobre las células vasculares del músculo liso, expresión elevada de mediadores del RAAS, alteraciones del metabolismo del calcio con desarrollo de hiperparatiroidismo complicando a los pacientes con el desarrollo de hipertrofia del ventrículo izquierdo, remodelado vascular e hipertensión arterial (HTA)6.
En modelos murinos con knockout del receptor de VitD (VDR) se observó una mayor incidencia de hipertensión, hipertrofia ventricular izquierda y aterosclerosis7, mientras que en animales con HTA (SHR), el empleo de VitD o análogos redujo la hipertrofia ventricular izquierda de manera equivalente al uso de losartán8. Por otro lado, en ratones normales, la deficiencia de VitD estimuló la expresión de renina, mientras que la inyección de la misma reduce su síntesis. Además, en estudios realizados en cultivos celulares se observó que esta relación se debería a un proceso inhibitorio de la VitD sobre la transcripción del gen de renina, a través de un mecanismo dependiente del VDR. Esto se confirmó debido a que los ratones que carecen del gen VDR desarrollan hiperreninemia, dando como resultado una elevada producción de angiotensinaII (AngII), lo que inexorablemente los llevó a hipertensión, hipertrofia cardíaca y aumento de la ingesta de agua8.
El bloqueo del VDR en células endoteliales de ratones mostró una respuesta alterada a la relajación dependiente del endotelio y exagerada a la infusión de AngII. Esta anomalía respondería —entre otros factores— al deterioro de la vía de señalización del óxido nítrico (NO). La respuesta cardiovascular estaría mediada por el efecto de la VitD sobre el endotelio, y su déficit conduciría a HTA9. También se observó que ratones diabéticos y knockout para VDR (VDR-KO) desarrollaron una nefropatía más grave que la de los ratones salvajes, lo que sugiere que la VitD protegería de la lesión renal mediante la regulación del RAAS10. En estrecha relación y con implicancias traslacionales, un metaanálisis realizado por Derakhshanian et al. sugirió que la VitD jugaría un papel nefroprotector, aunque los ensayos clínicos no han demostrado beneficios significativos por su uso, ni con análogos como el paricalcitol11. De interés, con relación a paricalcitol y metabolismo del calcio, el efecto de la VitD sobre la renina es independiente del metabolismo del calcio12. Más específicamente, Kong et al. trabajaron con un modelo de ratones transgénicos que sobreexpresan VDR humano para evaluar a las células productoras de renina y demostraron que la supresión de la expresión de renina por VitD in vivo es independiente de la hormona paratiroidea y del calcio13.
La activación inapropiada del RAAS ha sido reportada en ratones VDR-KO y 1α-hidroxilasa-KO14,15. Estos ratones desarrollaron HTA e hipertrofia miocárdica, que continuaron presentes incluso después de la normalización de la homeostasis del calcio; sin embargo, el bloqueo del RAAS con inhibidores de la enzima convertidora de la angiotensina normalizó la presión arterial y las alteraciones cardíacas. Por otro lado, el aumento de la activación del RAAS, la HTA y las alteraciones del miocardio pudieron ser tratadas exitosamente con VitD en ratones knockout para 1α-hidroxilasa. La activación de la renina con valores bajos de VitD se ha confirmado en estudios de cohorte en pacientes. Estos datos sugieren que los niveles plasmáticos de VitD pueden dar lugar a regulación positiva del RAAS16,17. En la actualidad, los efectos moleculares de la VitD sobre el RAAS son más claros debido al descubrimiento trascendental de que cuando VitD se une a la proteína de unión al elemento de respuesta de AMPc (CREB), impide la formación de un complejo CRE-CREB-CBP/p300 y este complejo suprime la expresión del gen de renina. Como resultado, la expresión de renina disminuye porque el CREB ya no puede unirse al elemento de respuesta al AMPc, y por lo tanto, no es capaz de activar la transcripción en la región promotora del gen de la renina18.
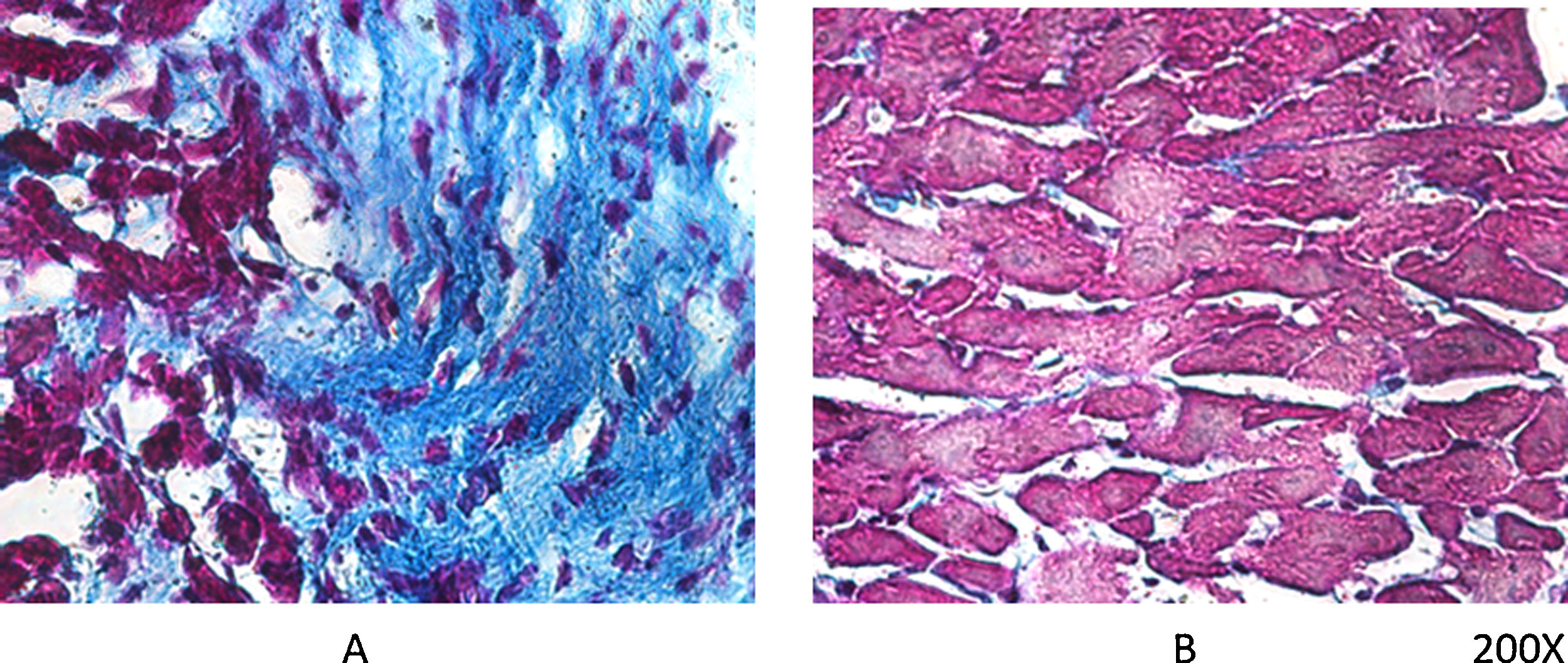
Según comentábamos previamente, los ratones VDR-KO muestran sobreexpresión de renina miocárdica y marcada hipertrofia de sus cardiomiocitos. Simpson et al. también demostraron que los animales deficientes en VitD tenían aumentada la incidencia de HTA, presentaban hipertrofia ventricular izquierda, y desarrollo de aterosclerosis19. En el ensayo PRIMO, que incluyó a 227 pacientes con enfermedad renal crónica estadios 3 a 4 y que fueron asignados al azar para recibir paricalcitol o placebo, el cambio en el índice de masa ventricular izquierda después de 12 meses no difirió entre los dos grupos20. Sin embargo, un estudio post hoc del ensayo PRIMO ha demostrado que 48 semanas de terapia con paricalcitol reducen significativamente el volumen auricular izquierdo, lo que se asociaría a reducción de la mortalidad21. Al respecto, nuestro laboratorio observó reducción en la expresión del VDR miocárdico en ratas con nefropatía obstructiva, lo cual podría estar relacionado con la remodelación del miocardio asociada con un aumento en la arritmogénesis. No obstante, la utilización de paricalcitol protegió contra estos cambios al restaurar los niveles de VDR miocárdico, reducir el proceso fibrótico y prolongar los potenciales de acción22 (fig. 2).
, mientras que en el mismo período de evolución pero con tratamiento diario de paricalcitol, a razón de 30ng/kg/día vía IP, se logró evitar el proceso fibrótico (B).")
Tricrómico de Masson en tejido miocárdico de rata. Luego de 60 días de evolución, la lesión miocárdica por nefropatía obstructiva se muestra claramente en (A), mientras que en el mismo período de evolución pero con tratamiento diario de paricalcitol, a razón de 30ng/kg/día vía IP, se logró evitar el proceso fibrótico (B).
Respecto a los mecanismos moleculares implicados en la enfermedad cardiovascular y el desacople del sistema RAAS, Klotho juega un papel protagónico. Su interacción ejerce una notable influencia en el desarrollo de enfermedades cardiovasculares en modelos animales de HTA, diabetes y enfermedad renal crónica. El factor Klotho es una proteína transmembrana involucrada en la regulación del metabolismo del calcio y del fosfato. Es producida en diversos tejidos y se expresa como forma soluble o anclada a la membrana plasmática. Inicialmente fue descubierta como una proteína con función antienvejecimiento. Se conocen tres tipos: α, β, y γ. Klotho ejerce su acción actuando como cofactor de una proteína crucial de la familia de los factores de crecimiento fibroblástico llamada factor de crecimiento fibroblástico 23 (FGF23). El FGF23 es una fosfatonina codificada en el locus 12p13 que promueve la excreción de fosfato en el túbulo contorneado proximal mediante la inhibición del cotransportador Na-Pi localizado en la membrana luminal. Su acción se desencadena luego de la unión al receptor FGFR1 solo si este se encuentra asociado a la proteína Klotho. Además, la interacción entre FGF23 y Klotho no solo regula la fosfatemia, sino que también disminuye la producción renal de VitD inhibiendo la enzima 1α-hidroxilasa. La VitD, a su vez, es capaz de incrementar la expresión de FGF23 y Klotho modulando la transcripción de sus genes. La regulación potencial del ARNm de klotho por VitD se dedujo de los estudios informados por Tsujikawa et al.23, quienes describieron el efecto de las manipulaciones farmacológicas y dietéticas de VitD en ratones sobre la expresión del mensajero klotho de 5,2kb24. Las alteraciones que condicionan reducción en la expresión y actividad de FGF23 y Klotho se asocian con aceleración de procesos de envejecimiento celular, calcificación vascular y de tejidos blandos, así como con mayor mortalidad, mientras que la normalización de los niveles de fosfato sérico revierte estos cambios patológicos25–27. De igual manera, se ha informado que los niveles bajos del cofactor Klotho se asocian a enfermedad cardiovascular, hipertrofia ventricular y mayor mortalidad28. La deficiencia genética del factor Klotho conduce a fibrosis cardíaca y desarrollo de hipertrofia ventricular izquierda. La disfunción del ventrículo izquierdo se detecta antes de que se presente hipertrofia y cambios fibróticos en el miocardio. Klotho es capaz de ejercer una contrarregulación de la acción profibrótica del TGFβ y la AngII. Las vías moleculares que intervienen en los procesos de hipertrofia y fibrosis son las mismas que conducen a estas alteraciones a nivel renal29. De central interés para la presente revisión es que los niveles de fosfato sérico impactan también en el metabolismo mitocondrial de forma significativa. El fosfato inorgánico regula la fosforilación oxidativa en varios niveles, lo cual se pone de manifiesto al observar que el incremento del fosfato extramitocondrial en el músculo cardíaco y esquelético porcino estimula en la mitocondria el consumo de oxígeno, incrementa los niveles de NADPH y regula el potencial transmembrana30. Además, el mayor influjo de equivalentes de reducción al complejo del citocromo III desencadenado por mayores niveles de fosfato origina mayor producción de especies reactivas del oxígeno (ROS) y como consecuencia aumenta el estrés oxidativo mitocondrial31. Esto constituye un eje relevante y señala el vínculo que se establece entre los niveles de fosfato inorgánico extramitocondrial, sus principales hormonas reguladoras —VitD, hormona paratiroidea (PTH), FGF23 y el cofactor Klotho—, el aparato cardiovascular, las ROS y el metabolismo mitocondrial.
La AngII, por su parte, al unirse al receptor AT1, conduce a un aumento en la producción de ROS por la activación de la enzima NADPH oxidasa31,32. También estimula la producción de NO, que en última instancia se combina con las ROS, particularmente con el anión superóxido O2− para formar peroxinitritos33. Esto conlleva mayor daño oxidativo y deteriora la disponibilidad del NO. En situaciones fisiopatológicas con hiperactivación del RAAS, como durante la enfermedad hipertensiva, la producción exacerbada de AngII asume un papel clave en la generación de ROS y contribuye al daño celular y tisular.
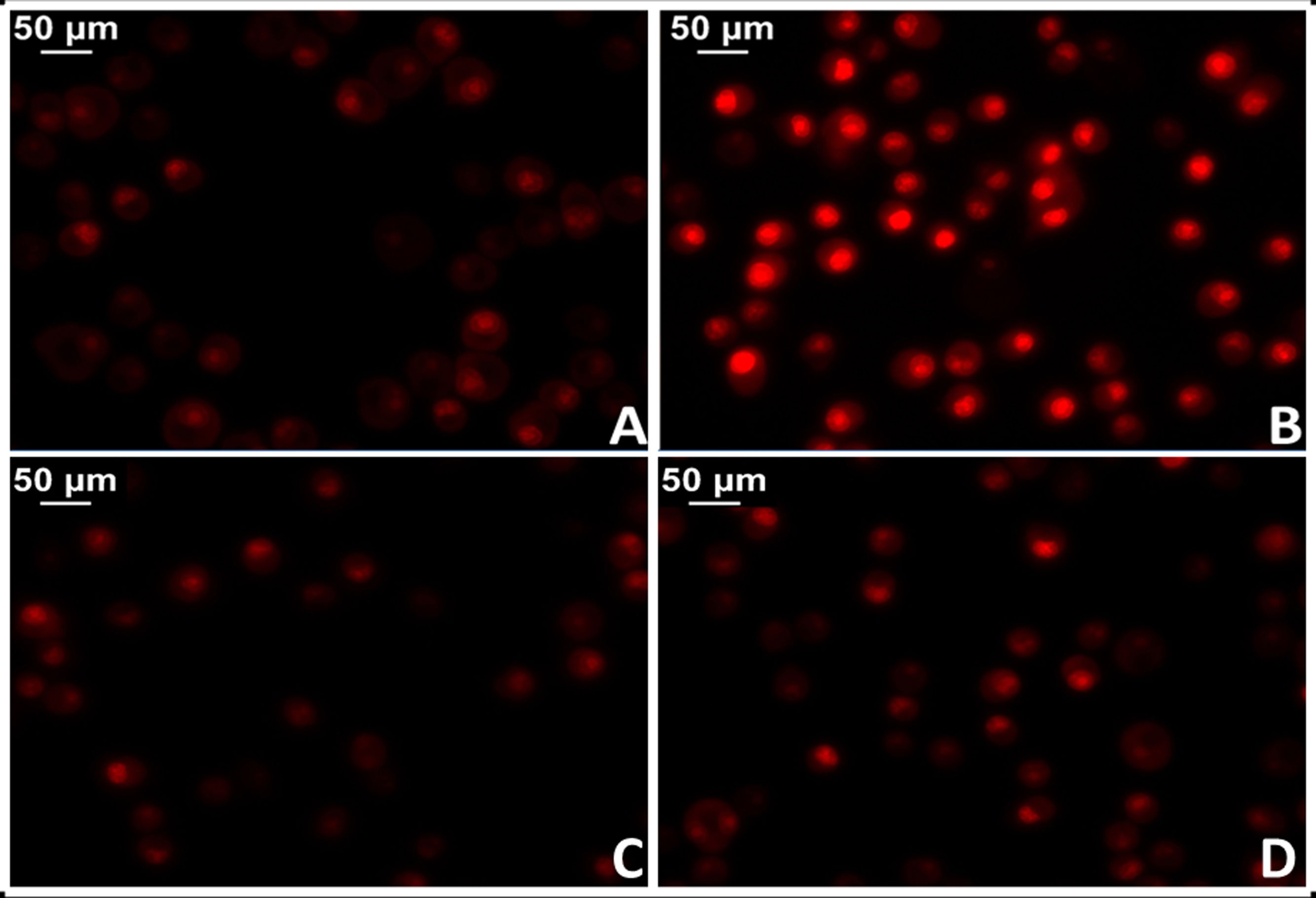
La administración de AngII provoca mayor oxidación proteica, mayor contenido de ADN mitocondrial y deleciones genéticas en las mitocondrias del músculo cardíaco de ratones, mientras que estos efectos dañinos no se manifiestan en modelos de ratones provistos de genes que sobreexpresan la catalasa mitocondrial (MCAT). Del mismo modo, en estos ratones la hipertrofia cardíaca fue menor34. En este sentido, se dispone de robustas evidencias que demuestran activación de la NADPH oxidasa mitocondrial por acción de la AngII e incremento en la producción de ROS mitocondrial35. En acuerdo, el bloqueo de AngII disminuye la producción de ROS y mejora la función mitocondrial. Más específicamente, la implementación de losartán en ratas espontáneamente hipertensas (SHR) logró prevenir la producción de H2O2, la actividad superóxido dismutasa mitocondrial y la citocromo oxidasa36,37. En este sentido, resultados preliminares de nuestro laboratorio en tejido miocárdico de ratas SHR tratadas con losartán demostraron una significativa reducción del daño mitocondrial con reducción de los niveles del anión superóxido (fig. 3).
 Tejido miocárdico proveniente de ratas normotensas (WKY). B) Tejido miocárdico proveniente de ratas espontáneamente hipertensas (SHR). C) Tejido miocárdico proveniente de ratas normotensas tratadas con losartán a razón de 40mg/kg/día en el agua de bebida (WKY+Los). D) Tejido miocárdico proveniente de ratas espontáneamente hipertensas tratadas con losartán a razón de 40mg/kg/día en el agua de bebida.")
Determinación del estrés oxidativo celular en tejido miocárdico de rata mediante la técnica del colorante fluorescente dihidroetidio para evaluar la producción de anión superóxido. A) Tejido miocárdico proveniente de ratas normotensas (WKY). B) Tejido miocárdico proveniente de ratas espontáneamente hipertensas (SHR). C) Tejido miocárdico proveniente de ratas normotensas tratadas con losartán a razón de 40mg/kg/día en el agua de bebida (WKY+Los). D) Tejido miocárdico proveniente de ratas espontáneamente hipertensas tratadas con losartán a razón de 40mg/kg/día en el agua de bebida.
Sobre el metabolismo mitocondrial se ha descrito que VDR inhibe la cadena respiratoria. El silenciamiento de VDR mejoró la actividad respiratoria de las células. La inducción de VDR reduce la transcripción de proteínas de la cadena respiratoria y el potencial de membrana mitocondrial, desviando el metabolismo y optimizando el consumo de energía, mientras que la pérdida o reducción en la expresión de VDR da como resultado una inhibición importante del crecimiento celular, con acumulación en las fases G0 y G1 del ciclo y reducción de la fase M. Los efectos del silenciamiento de VDR sobre la proliferación se confirmaron en varias líneas celulares de cáncer humano. Una característica distintiva de las células tumorales es el metabolismo alterado que apoya la rápida proliferación celular, siendo la mitocondria responsable de muchos procesos metabólicos alterados y que aumentan la proliferación celular38. Paralelamente, VDR participa en la modulación metabólica mitocondrial en tejido graso. Específicamente, a nivel del tejido graso la acción mitocondrial de la VitD se ejerce sobre el metabolismo de los ácidos grasos. En estudios realizados en ratas transgénicas, la sobreexpresión de VDR llevó a una disminución de la expresión de proteínas desacoplantes, una disminución de beta-oxidación y lipólisis. Este desacople metabólico se vio acompañado de la disminución de genes codificantes para enzimas clave en dichas vías de señalización, tales como hexoquinasas, carnitil-palmitoil transferasa y lipasa. En dichos ratones se observó un aumento de la ganancia de tejido adiposo, acompañado con niveles bajos de leptina y altos niveles de colesterol. Como contraparte, en ratones VDR-KO se observó un aumento en la expresión de las proteínas desacoplantes en tejido adiposo pardo, junto con un aumento de la expresión de las mismas en adipocitos pardos tratados con VitD, lo que sugiere una acción inhibitoria de VitD sobre las proteínas desacoplantes y la disminución de la utilización de lípidos, con una redirección del acetil-CoA hacia vías biosintéticas39.
En relación con conceptos previos, uno de los efectos genómicos de la VitD consiste en disminuir la producción de prorrenina mediante la inhibición de su transcripción, lo que conduce a menor activación del RAAS. La AngII reduce la expresión de Klotho, y esto hace que la relación FGF23/Klotho aumente, lo que condiciona una reducción en los niveles de VitD y que además impacte negativamente en la regulación del sistema RAAS, reforzando su activación40. Por lo tanto, se puede concluir que la VitD interviene decisivamente en el control del desequilibrio de la oxidorreducción inducido por la hiperactivación del RAAS.
Otro mediador molecular que juega un rol protagónico relacionado con la VitD son las denominadas sirtuinas. Al respecto, la VitD ha demostrado reducción del estrés oxidativo mediante el incremento en la expresión de sirtuina 1 (SIRT1) y AMPK41. La familia de las sirtuinas comprende un conjunto de 7 proteínas involucradas en la modulación de los procesos vinculados al desarrollo de enfermedades asociadas al envejecimiento y ejerce múltiples funciones en el metabolismo y la proliferación celular26. Básicamente son enzimas con actividad desacetilasa que dependen de los niveles intracelulares de NAD+. La interrelación entre las sirtuinas y VitD se lleva a cabo de manera dual debido a que estimulan de forma recíproca su expresión en un ciclo de retroalimentación positiva42. Más específicamente, la sirtuina 3 (SIRT3) modula el funcionamiento del ciclo de la urea y la oxidación de los ácidos grasos en la matriz mitocondrial. También ejerce una influencia protectora en la mitocondria en virtud de su capacidad para crear un ambiente reductor mediante el aumento de la actividad de la enzima glutatión reductasa, lo cual previene los efectos dañinos del estrés oxidativo. De esta manera, la relación entre el glutatión reducido disponible y el glutatión oxidado derivado de la reducción de especies nocivas se incrementa en favor del primero43. Al respecto, se ha demostrado que la hiperactivación del RAAS con elevación de AngII —como aquella presente en enfermedades cardiovasculares— altera las funciones de SIRT3 inhibiendo su expresión44. Por otro lado, SIRT1 incrementa la expresión de la enzima convertidora de angiotensina tipo II (ACEII). Al respecto, se conocen ampliamente los efectos antiinflamatorios, antiproliferativos y vasodilatadores de los productos escisionales de la ACEII sobre el perfil fisiopatológico de las enfermedades cardiovasculares. El procesamiento diferencial de los precursores de angiotensina mediante la vía gobernada por ACEII produce la angiotensina 1-7 (Ang 1-7) capaz de activar un receptor Mas (MasR) que ejecuta sus efectos en los órganos blanco. De interés, muy recientemente se ha demostrado en ratas hipertensas que el uso de un antagonista específico de MasR anuló las acciones antioxidantes e inmunomoduladoras de VitD. Estos hallazgos indican la participación de la vía ACEII/Ang1-7/MasR en los mecanismos protectores de la VitD45.
Todos estos datos siguen aportando evidencia sobre los efectos no genómicos de VitD, sumados al clásico mecanismo donde VDR actúa como un factor de transcripción que migra al núcleo a través de los poros de la membrana nuclear. Sin embargo, recientemente se ha sugerido un nuevo mecanismo de acción de VitD relacionado con la localización intramitocondrial de sus receptores VDR39.
Al respecto, se ha demostrado en plaquetas humanas la presencia del receptor de VitD en sus mitocondrias utilizando anticuerpos anti-VDR. Las plaquetas constituyen una muestra ideal para el estudio de la localización extranuclear del VDR puesto que carecen de núcleo46. En este sentido se avanzó, y en modelos experimentales con inducción del RAAS y sobreexpresión del receptor AT1 se pudo demostrar que el empleo de paricalcitol causó reducción de la expresión AT1 e inducción del VDR intramitocondrial, lo cual sugiere un vínculo citoprotector directo entre paricalcitol y el receptor AT1 mediado por la activación del VDR mitocondrial47. Posteriormente, nuestro laboratorio propuso que esa disminución de AT1 inducida por VDR podría ser una consecuencia de la protección celular mediada por la proteína de shock térmico 70 (Hsp70)48. De hecho, observamos en las mitocondrias una asociación significativa en la expresión de AT1, VDR y Hsp70. Previamente a nuestros hallazgos, se determinó que Hsp70 interactúa con el VDR y juega un papel en el control de su concentración intracelular49. También se ha demostrado en riñón de rata que VitD es un inductor de Hsp7050. En estrecha relación, se pudo establecer, en un modelo de afectación cardiovascular con insuficiencia renal, una significativa prevención del daño renal y remodelación miocárdica asociado a reducción en el estrés oxidativo, fibrosis y apoptosis con la reducción de AT1 e incrementos de Hsp70 y VDR51.
Al respecto cabe mencionar que previamente se demostró —en células embrionarias de riñón de rata, provenientes de muestras de tumor primario de Wilms, así como en células cultivadas con expresión inducible de la proteína del tumor de Willms 1 (WT-1)— asociación entre Hsp70 y WT-152. Esta proteína es necesaria para procesos nefrogénicos y cardiogénicos. De interés, se ha descrito que Hsp70 es un cofactor importante para la función de WT-1, y se sugirió un papel potencial para esta chaperona durante el proceso de nefrogénesis. Además, han sido identificados mensajero y proteína del receptor de VDR en los riñones de embriones, lo que sugiere un papel del sistema de la VitD en el desarrollo renal. Los patrones de expresión temporal de genes WT-1 y VDR correlacionan estrechamente en el riñón durante el desarrollo. Adicionalmente se ha demostrado que los promotores de los genes VDR humanos y de ratón contienen varios sitios consenso de unión a WT-153,54.
Estudios clínicos y epidemiológicosLa prevalencia de HTA en el año 2000 fue del 24,6% en todo el mundo, y con base en proyecciones se estima que alcanzará el 29,2% de la población en 202555. La HTA es una enfermedad prevalente que afecta principalmente al corazón, los riñones y el cerebro. Su mejor manejo y nuevos tratamientos podrían disminuir el daño sobre estos órganos blanco. Sobre mecanismos y posibles nuevos enfoques, un metaanálisis previo al 2015 que incluyó 8 estudios prospectivos informó que el nivel de VitD se asoció inversamente con la incidencia de HTA (RR: 0,70; IC del 95%: 0,58 a 0,86)56. Esto fue objetivado en estudios clínicos llevados a cabo en las últimas dos décadas y que han resaltado una asociación inversa entre la concentración plasmática de 1,25(OH)2D3, la presión arterial y/o la actividad de la renina plasmática, tanto en varones normotensos como en pacientes con hipertensión esencial16,57.
Algunos metaanálisis más recientes observan estudios con resultados positivos en el tratamiento de la presión arterial. Tomando en cuenta el punto de corte de 20ng/mL (o 50nmol/L) como déficit de VitD, se observaron resultados favorables con la suplementación de VitD a personas obesas (IMC mayor de 30kg/m2) e hipertensas de más de 50 años con niveles deficitarios de VitD inferiores a 20ng/mL (50nmol/L)58. Esto se asocia con la disminución de la absorción de VitD en personas de edad avanzada.
En una muestra de población adulta, Sabanayagam et al. encontraron que los niveles más bajos de VitD sérica estaban asociados con prehipertensión, con un riesgo relativo de 1,48 (IC: 1,16-1,90)59. La deficiencia de VitD como factor de riesgo ambiental favorece el aumento del tono vascular, que puede no desempeñar un papel importante en la regulación de la homeostasis normal de la presión arterial, pero sirve como desencadenante para contribuir al desarrollo de HTA en personas vulnerables de mediana edad60.
Los estudios experimentales en ratones han indicado que la suplementación con VitD reduce significativamente la síntesis de renina y la presión arterial. Es posible y concebible que mecanismos similares de acción puedan encontrarse en seres humanos. Sin embargo, aún no se han obtenido los resultados esperados. No obstante, en línea con los datos antes mencionados, ciertos estudios han demostrado una reducción de la presión arterial en pacientes con hipertensión primaria que recibían suplementos de VitD61, y una reducción de la presión arterial, renina plasmática y niveles de AngII en pacientes con hiperparatiroidismo secundario62,63. El tratamiento con VitD logró una reducción del 50% en el ARNm de renina, en comparación con el grupo control. Estos hallazgos sumados a los resultados experimentales implican la importancia de la VitD como un supresor efectivo de la síntesis de renina64. Además, otros ensayos han demostrado la mayor efectividad del tratamiento con paricalcitol en comparación con suplementos de VitD. Entre sus beneficios se ha observado: reducción de niveles de renina, disminución de interleucinas 1 y 6, factor de necrosis tumoral y valores de proteinuria demostrando un efecto nefroprotector65.
Queda así expuesto también el protagonismo de la vía inflamatoria y la señalización mediada por VitD en la enfermedad cardiovascular. De especial interés, la señalización del VDR en leucocitos y macrófagos cumple un rol importante en el desarrollo de aterosclerosis y HTA. Al menos parte del mecanismo antiaterosclerótico consiste en bloquear la activación del RAAS local en los macrófagos y dentro de la lesión aterosclerótica66.
En el marco del desarrollo de la aterosclerosis, la afectación de la aorta y su pared condiciona la aparición de los aneurismas aórticos, y en estrecha relación con el tema central de la presente revisión, los aneurismas aórticos están fuertemente asociados a la HTA y el sistema RAAS, como lo demuestran estudios previos. Específicamente, la HTA determina la aparición de enfermedades de la pared arterial, como ateromas, disecciones y aneurismas. Estos últimos, importantes en relación con su localización y gravedad. El aneurisma aórtico se caracteriza por degeneración progresiva de la estructura de la pared arterial por inflamación crónica, aterosclerosis y factores inherentes que determinan cambios con remodelación de la pared y que conducen a la dilatación67. La dilatación a medida que progresa se torna irreversible y la rotura puede ser clínicamente fatal. Así, algunos pacientes sobreviven al cuadro mientras que en otros se presenta como muerte súbita. De particular interés, las alteraciones provocadas se han relacionado también con el déficit de VitD y la progresión de la HTA en su evolución natural. Esto fue evidenciado en un seguimiento de pacientes con aneurisma versus controles (revisiones y metaanálisis)68–70.
En cuanto a los posibles mecanismos moleculares responsables, existe evidencia que sugiere una relación estrecha entre la VitD y la regulación del sistema RAAS, como previamente fue mencionado. Más específicamente, se utilizaron ratones como modelo de aterosclerosis por deficiencia de apolipoproteína E (ApoE−/−) y donde además se les indujo la formación de aneurismas de aorta abdominal por administración de AngII. Como resultado relevante se encontró que el tratamiento oral con calcitriol redujo la formación de aneurismas con un marcado aumento en la interacción VDR-RXR-α (receptor X retinoide α) en las aortas de ratones tratados con calcitriol. Además, exhibieron una menor infiltración de macrófagos, metaloproteinasa de matriz (MMP) y expresión de quimiocinas en las paredes aórticas suprarrenales, al parecer, como consecuencia de la activación de las interacciones VDR-RXR-α71. Este estudio proporcionó sólida evidencia sobre que el tratamiento crónico con calcitriol reduciría la formación de aneurisma inducida por exaltación del RAAS debido a que la AngII es uno de sus principales exponentes. Estos efectos estarían asociados con una respuesta inflamatoria reducida y regulación de la homeostasis de la matriz extracelular a través de la heterodimerización VDR-RXR-α. A la luz de estos resultados, la activación de VDR podría representar un objetivo terapéutico prometedor para el tratamiento del aneurisma aórtico71.
Paralelamente, los efectos ejercidos por la activación del VDR cumplen una función crítica en la modulación de las señales que intervienen en los procesos ateroscleróticos. En las lesiones ateroscleróticas incipientes, la diferenciación de linfocitos T hacia el fenotipo Th1 impulsa la activación de macrófagos mediante la producción de IFNγ. La fagocitosis de partículas de LDL modificadas y su acumulación en la región subintimal de la pared arterial forma, en última instancia, la estría adiposa precursora de las lesiones ateroscleróticas establecidas. La VitD es capaz de interferir con este proceso patológico a través de la regulación de la diferenciación de los linfocitos T cooperadores. Al respecto, se ha observado que los análogos de VitD inducen el cambio hacia un fenotipo Th2 en los linfocitos T en presencia de la acción de IL 4. A su vez, los análogos de VitD poseen un efecto inhibitorio en la producción de IFNγ, lo cual lleva a menor diferenciación de linfocitos Th1 y menor reclutamiento de células fagocíticas72. El aumento de la acción del VDR también suprime el ingreso de partículas LDL oxidadas hacia la íntima arterial y disminuye la formación de células espumosas73. Como aspecto fundamental y previamente citado, la acción de VitD en el proceso antiaterosclerótico parece depender también de la menor activación del sistema RAAS local en la placa ateromatosa. Esto es debido a que AngII, a través de la ocupación de los receptores AT1, promueve la expresión de genes proinflamatorios e incrementa el estrés oxidativo mitocondrial como consecuencia de la activación crónica de la vía del NFκB y la enzima NADPH oxidasa74.
Paralelamente, la aterosclerosis como enfermedad crónica se asocia con disfunción cardiovascular que incluye infarto de miocardio, angina inestable, muerte cardíaca súbita, accidente cerebrovascular, aneurismas en aorta y trombosis periféricas. Sobre aterosclerosis, se ha pronosticado que será una de las principales causas de muerte en el mundo para 2020 motivado por una clara lesión endotelial debida al estrés oxidativo asociado con factores de riesgo cardiovascular que incluyen diabetes e HTA, entre los más destacados75.
Al respecto, Krause et al.76 realizaron un estudio en el que los participantes fueron expuestos a la radiación UVB en una cama solar 3 veces por semana durante 3 meses. Los sujetos experimentaron un aumento del 180% en sus niveles de VitD y una reducción de 6mmHg tanto en presión arterial sistólica como diastólica. Luego, un pequeño estudio aleatorizado, controlado contra placebo, en pacientes con diabetes tipo 2 (DM2) y con niveles basales bajos de VitD, mostró que una sola dosis de 100.000UI de VitD redujo la presión arterial sistólica en 14mmHg, y la misma dosis mejoró significativamente la función endotelial medida por el flujo sanguíneo del antebrazo77. En el estudio NHANES3, la presión arterial sistólica media fue aproximadamente 3mmHg menor en los individuos en el quintil más alto de la VitD sérica en comparación con los del quintil más bajo78. Sin embargo, y a pesar de que varios estudios epidemiológicos han demostrado que puede haber una asociación entre hipertensión y bajos niveles de VitD, los resultados no avalan aún el uso de VitD o sus análogos como tratamiento individual para pacientes con hipertensión o como una intervención a nivel poblacional para reducir la presión arterial79.
Scragg et al.78 publicaron sus hallazgos sobre la relación entre los niveles de VitD y la presión arterial. Los autores estudiaron los datos del NHANES3 y encontraron una significativa relación inversa entre los niveles de VitD y los valores de presión arterial, que era evidente incluso después de ajustar por variables como edad, sexo, etnia y actividad física. Judd et al.80 también analizaron datos del NHANES3 y mostraron una relación inversa estadísticamente significativa entre las concentraciones de VitD circulantes y la presión arterial sistólica. Martins et al.81, también sobre datos del NHANES3, encontraron que un nivel bajo de VitD estaba asociado con un mayor riesgo de padecer HTA. Estos resultados condicionarían posibles explicaciones para justificarlos y así se desarrollaron los estudios sobre el Riesgo Cardiovascular Ludwigshafen y de la Salud (LURIC). Aquí se tuvo como objetivo documentar una posible asociación entre los distintos tipos de VitD y RAAS circulantes en una gran cohorte de pacientes (n=3.316) con derivación para angiografía coronaria. Después de medir 25(OH)D3, 1,25(OH)2D3, renina plasmática y concentración de AngII, lograron demostrar por vez primera en seres humanos, una asociación independiente entre ellos82. Sin embargo, previamente también se evaluó la asociación en la reducción de la HTA y la suplementación de VitD. Así, en un metaanálisis de estudios entre 1966 y 2014 se concluyó que la presión arterial no bajó con este tratamiento y no descendió en forma significativa, por lo que no se recomendaría su utilización como antihipertensivo83.
Por lo tanto, y frente a tantas controversias y con el ánimo de investigar en detalle la posible relación presión arterial versus niveles de VitD, hoy se encuentra en desarrollo el estudio VITAL cuyos objetivos primarios son determinar si la suplementación con VitD y aceite de pescado disminuye la presión arterial ambulatoria de 24h en comparación con el placebo en una subcohorte de 1.000 participantes; además se busca evaluar si la suplementación con VitD y aceite de pescado reduce el riesgo de hipertensión incidente en comparación con el placebo entre todos los participantes VITAL aleatorizados sin hipertensión basal; y si la suplementación con VitD y aceite de pescado cambia favorablemente los biomarcadores relacionados con hipertensión en comparación con el placebo. También se invitará a una subcohorte representativa de 1.000 participantes del estudio VITAL sin hipertensión de las principales áreas metropolitanas seleccionadas de los Estados Unidos a participar en visitas de estudio en sus hogares al inicio del estudio y tras 2 años de seguimiento. Durante estas visitas, se les pedirá a los participantes que usen monitores para registrar las mediciones de presión arterial ambulatoria de 24h, proporcionar sangre en ayunas, muestras de orina puntuales y otras mediciones clínicas. Los resultados se esperan luego del cierre del estudio, que finalizaría en noviembre del 2020. La concreción de estos estudios proporcionará evidencia más relevante para apoyar o refutar los posibles roles preventivos de la VitD y los ácidos grasos omega-3 en la presión arterial y el desarrollo de la hipertensión84.
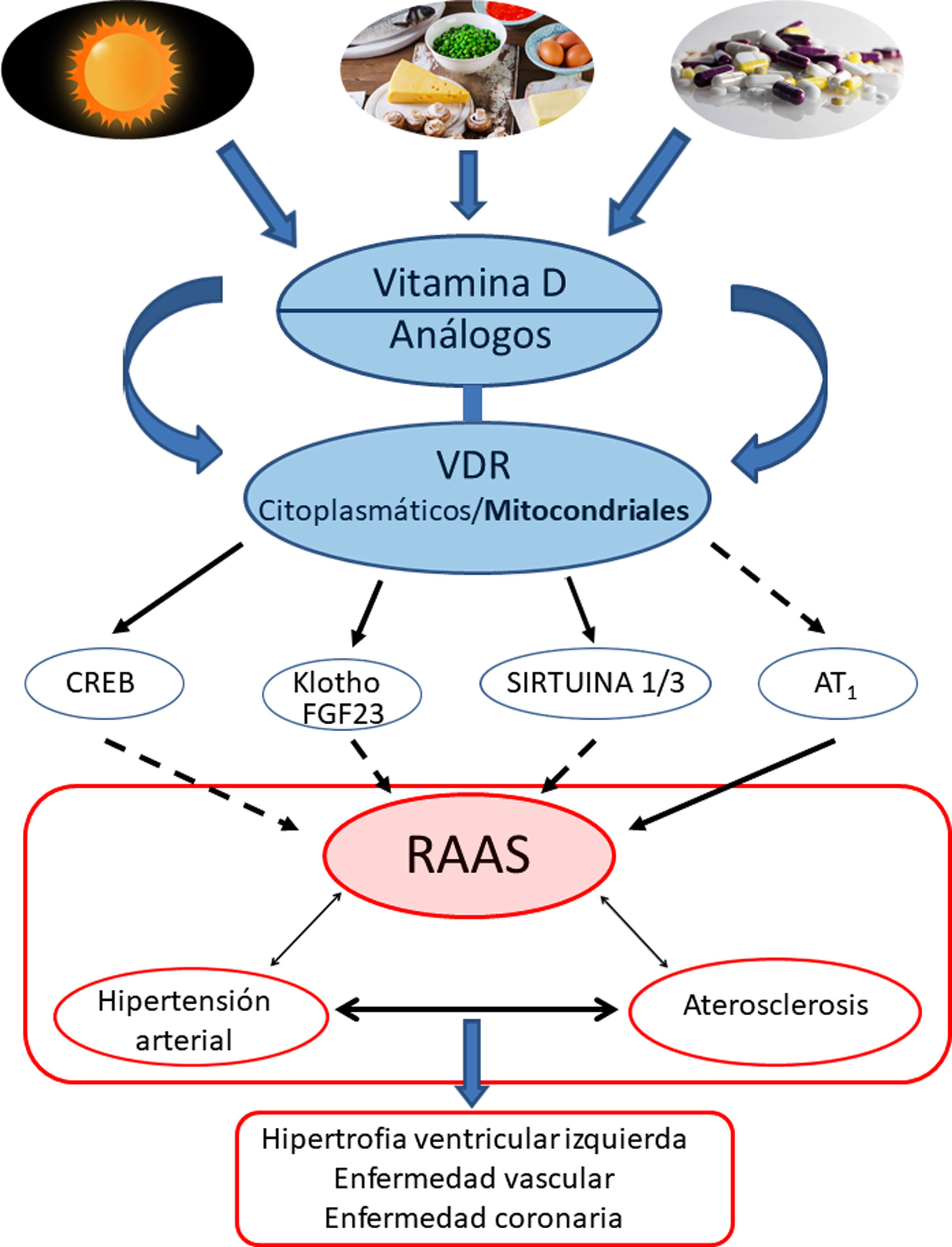
Conclusiones y perspectivasEl protagonismo de la VitD y su efecto protector contrapuesto a la HTA estaría respaldado fundamentalmente por sus efectos regulando negativamente la vía del RAAS. Más específicamente, los VDR modularían a nivel mitocondrial la cadena respiratoria, lo cual influiría en el remodelado cardiovascular. La activación de los VDR reduciría el daño oxidativo y preservaría la viabilidad celular y tisular. Los estudios en animales han demostrado claramente que la deficiencia de VitD se asocia a HTA, aterosclerosis e hipertrofia cardíaca (fig. 4).

Resumen gráfico/esquemático de las principales vías de señalización implicadas en la modulación de vitamina D —y sus receptores tanto citoplasmáticos como mitocondriales— sobre el sistema RAAS y su impacto en el desarrollo de la enfermedad cardiovascular y sus principales exponentes representados aquí como hipertensión arterial, aterosclerosis y la enfermedad coronaria. Las fechas continuas indican inducción/estimulación, mientras que las flechas discontinuas indican inhibición.
En contraposición, los ensayos clínicos muestran resultados contradictorios y no categóricos, que en parte pueden atribuirse a su diseño. Son por ello deseables estudios que establezcan el rol de la VitD en el marco del tratamiento actual para la HTA. Es importante llegar a establecer las acciones y resultados al tratar con VitD a los pacientes en las distintas etapas de la enfermedad hipertensiva, para volcar en ellos los beneficios comprobados en los estudios de investigación básica.
Los receptores VDR se encuentran presentes en músculo liso vascular, endotelio, células musculares cardíacas e inclusive dentro de la principal organela responsable —cuando se encuentra disfuncional— de la enfermedad hipertensiva cardiovascular, la mitocondria. Estudios observacionales han demostrado una relación entre los niveles bajos de VitD, presión arterial e inclusive calcificaciones de las arterias coronarias. Los niveles bajos de VitD se asocian con mayor carga oxidativa, marcadores inflamatorios y daño mitocondrial. Algunos estudios en animales y humanos han indicado que la VitD podría tener un papel clave en la homeostasis de la glucosa y el desarrollo de diabetes.
Finalmente, la concreción de estudios como el VITAL podría clarificar aspectos aún no comprendidos sobre la intrincada red de señalización, que resultan simples en ensayos experimentales pero complejos en los estudios clínicos, para apoyar o refutar los posibles beneficios de la VitD y la enfermedad cardiovascular hipertensiva.
FinanciaciónEste trabajo ha contado con el apoyo financiero proporcionado por subsidios del Consejo de Investigación y Tecnología de la Universidad de Cuyo (SECyT), Mendoza, Argentina, y de ANPCyT FONCyT, ambas adjudicadas a Walter Manucha. Subsidio n.o PICT 2016-4541.
AutoríaTodos los autores contribuyeron de igual manera en la concepción y diseño de la revisión, con una contribución sustancial sobre los datos, análisis e interpretación de los contenidos, redacción y revisión crítica del artículo para su contenido intelectual.
Conflicto de interesesLos autores declaran que no hay conflictos de interés potenciales con respecto a la investigación, autoría y/o publicación de este artículo.