



Posterior tibial tendon dysfunction is a common cause of adult flat foot deformity, and its etiology is unknown.
PURPOSEIn this study, we characterized the morphologic pattern and distribution of types I, III and V collagen in posterior tibial tendon dysfunction.
METHODTendon samples from patients with and without posterior tibial tendon dysfunction were stained by immunofluorescence using antibodies against types I, III and V collagen.
RESULTSControl samples showed that type V deposited near the vessels only, while surgically obtained specimens displayed type V collagen surrounding other types of collagen fibers in thicker adventitial layers. Type III collagen levels were also increased in pathological specimens. On the other hand, amounts of collagen type I, which represents 95% of the total collagen amount in normal tendon, were decreased in pathological specimens.
CONCLUSIONFibrillogenesis in posterior tibial tendon dysfunction is altered due to higher expression of types III and V collagen and a decreased amount of collagen type I, which renders the originating fibrils structurally less resistant to mechanical forces.
Posterior tibial tendon dysfunction (PTTD) is one of the most common causes of acquired flat foot deformity in adults, and results in significant morbidity due to the pain and development of secondary osteoarthritis.1,2 Multifactorial etiology has been proposed to explain tendon degeneration in PTTD, including trauma, anatomic disorders and mechanical factors as well as inflammatory and ischemic processes.3 It was previously thought to be secondary to an inflammatory process resulting in chronic tendinitis, but recent histopathologic studies revealed a degenerative tendinosis with non-specific reparative response and marked disruption of the linear orientation of collagen bundles, generating the term tendinopathy to describe this clinical condition.4,5
Recent biochemical studies have demonstrated a disproportion of several types of collagen in PTTD. Normal tendons are characteristically composed of more than 95% of type I collagen, with relatively small amounts of types III, IV and V collagen. However, a higher proportion of types III and V collagen in PTTD has been described, which may contribute to a decrease in the mechanical resistance of the tissue because types III and V collagen build thinner fibers than collagen type I.6 Our hypothesis is that this difference in terms of collagen types could be an adaptive remodeling of the tendon. Thus, our objectives were to investigate the histoanatomical distribution of collagen types I, III and V in the posterior tibial tendon by immunofluorescence and study its influence in tendon rupture.
MATERIALS AND METHODSTendon acquisitionPathologic group: The sample consisted of nine female patients with an average age of 53 years (range: 41 to 69). All patients presented with persistent and disabling symptoms of pain lasting at least six months, swelling on the medial aspect of the foot and foot planovalgus deformity with varying degrees of hind foot valgus and fore foot abduction with loss of longitudinal arch. Surgical treatment included resection of the diseased portion of posterior tibial tendon, flexor digitorum longus transfer to the navicular bone and medial calcaneal sliding osteotomy. All surgical tendon sections were analyzed by morphological study in both groups.
Control group: The control group consisted of three women. The average age of the patients at the time of surgery was 49 years (range: 29 to 72). Three pantalar arthrodeses were performed. All operations were performed as salvage procedures to treat painful osteoarthritis or deformity, involving both the ankle and subtalar joints following post-traumatic injury. Additionally, to identify the normal pattern collagen histoarchitecture, these tendons were also compared to apparently normal tendon (n=3) obtained in necropsies of patients who died from non-articular causes.
Histological studyThe samples were fixed in 10% buffered formalin, decalcified, embedded in paraffin and sectioned for microscopic examination. Histologic fragments were stained with Hematoxylin and Eosin (H&E) and Masson’s trichrome and analyzed under a light microscope. Sections were searched for abnormalities in collagen bundle orientation, vascularization and inflammation.
ImmunofluorescenceImmunofluorescence was used for collagen identification in 3 mm paraffin-embedded sections mounted on methacryloxypropyltri-methoxysilane (Sigma Chemical Co.; St. Louis, Missouri, USA) slides, in a manner analogous to a previously described procedure.7 The sections were dewaxed in xylene and rehydrated in graded ethanol. Antigen retrieval was done by enzymatic treatment of tendons with bovine pepsin (10000 UTD, Sigma Chemical Co.; St. Luois, Missouri, USA) in acid buffer (0.5 M) (2 mg/ml) for 30 min at 37°C, and subsequent incubation with 5% milk in phosphate buffer pH 7.0 were performed. Next, the slides were incubated with either polyclonal rabbit anti-human calf type I (1:200 dilution) or anti-human placenta type V (1:3500 dilution) collagen8 or mouse monoclonal anti-human type III collagen (1:100 dilution) (Oncogene; San Diego, USA). Incubation occurred overnight at 4oC in a humid atmosphere. The sections were then incubated with a FITC-conjugated anti-rabbit or mouse immunoglobulin (1:50 dilution, Sigma Chemical Co.; St. Luois, Missouri, USA) as a secondary antibody for 60 minutes and mounted with an aqueous mounting medium.
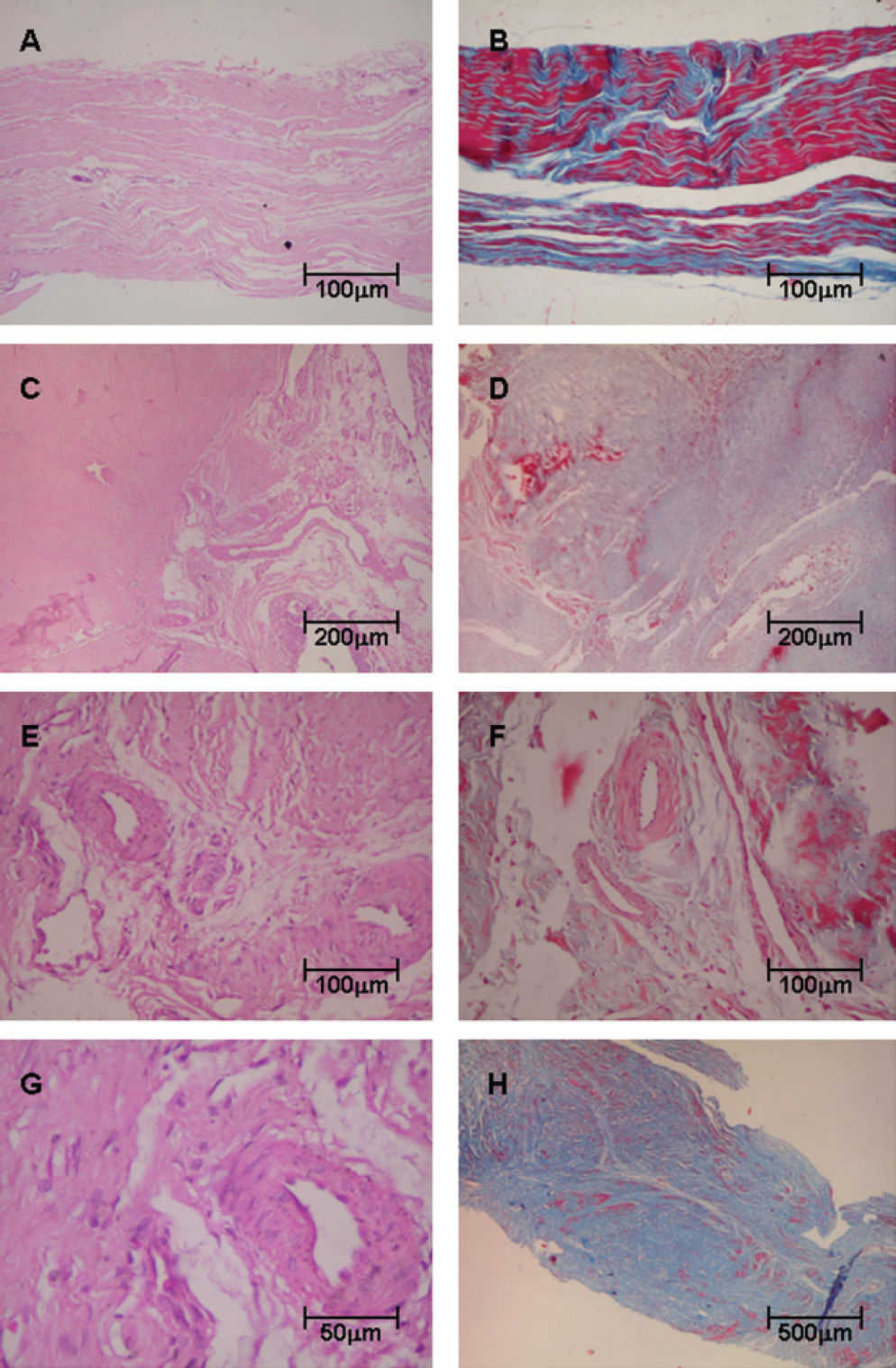
RESULTSHistopathologic analysis showed that control tendons displayed normal architecture characterized by parallel or linear orientation of collagen bundles, low vascular degree and scant fibroblast proliferation (Figure 1, panels A and B). In contrast, the posterior tibial tendon specimens from patients with PTTD demonstrated remarkable distortion of the architecture, which was modified by large pale areas with disruption of the normal linear orientation of collagen bundles and characterized by displaying fibrils in a wavy pattern. In PTTD, vessels were dilated and markedly increased in number (Figure 1, panels C and D). A subset of these increased vessels had complex branching that was caused by the proliferation of plump fibroblasts embedded in a myxoid stroma (Figure 1, panels E, F and G). In panel H, the final result of the remodeling process of tendonitis in PTTD is shown.
 patients, stained with Hematoxylin & Eosin (Panels A, C, E, G) and Masson’s trichrome (Panels B, D, F, H). Control tendons display normal architecture with parallel or linear orientation of collagen bundles and a low degree of vascularization (A) and (B). In contrast, (C) and (D) show tendon specimens from PTTD patients with architecture distortion and large pale areas with a wavy pattern of collagen bundles. In panels (E), (F) and (G), tendons from PTTD patients show increased vessels with complex branching caused by proliferation of fibroblasts and myxoid stroma. In (H), the final results of the remodeling process of tendonitis in PTTD are shown. (Original magnification: X 40 in panel H; X 100 in panel A, B, C, D and H; X 200 in panels E and F; X 400 in panels G)")
Tendon samples obtained from control and posterior tibial tendon dysfunction (PTTD) patients, stained with Hematoxylin & Eosin (Panels A, C, E, G) and Masson’s trichrome (Panels B, D, F, H). Control tendons display normal architecture with parallel or linear orientation of collagen bundles and a low degree of vascularization (A) and (B). In contrast, (C) and (D) show tendon specimens from PTTD patients with architecture distortion and large pale areas with a wavy pattern of collagen bundles. In panels (E), (F) and (G), tendons from PTTD patients show increased vessels with complex branching caused by proliferation of fibroblasts and myxoid stroma. In (H), the final results of the remodeling process of tendonitis in PTTD are shown. (Original magnification: X 40 in panel H; X 100 in panel A, B, C, D and H; X 200 in panels E and F; X 400 in panels G)
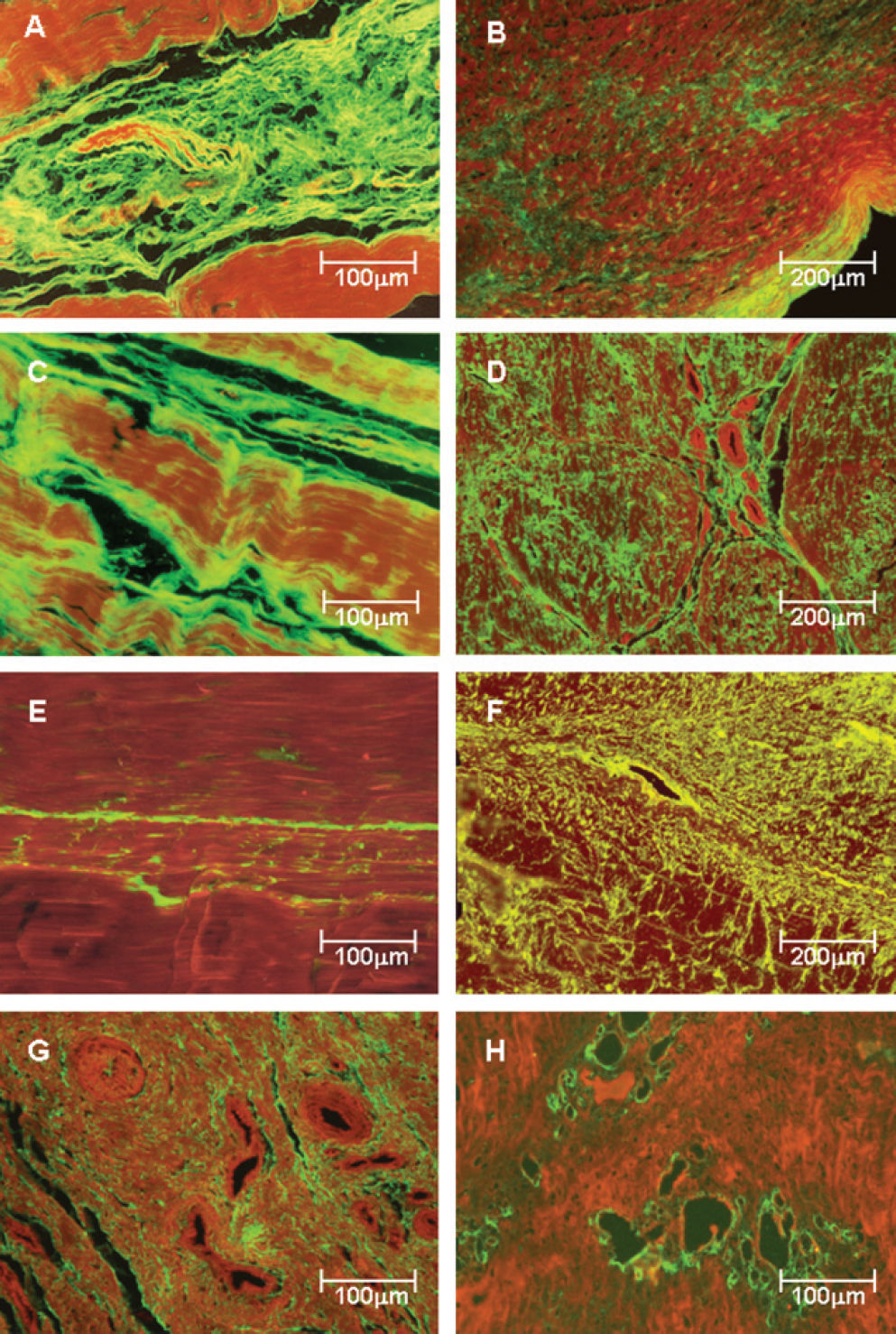
Collagen Expression in Posterior Tibial Tendon Matrix Types I, III and V collagen fibers in control (Figure 2 -panels A, C and E) and pathologic samples (Figure 2 - panels B, D, F, G and H) were immunostained. Clearly, tendons from the control patients show a green birefringence for type I, III and V collagens. Type I collagen presents a parallel orientation of the fibers in all structures of the tendon (Figure 2A), whereas type III physically interact with the type I collagen fibers and the corresponding vessels in a homogeneous pattern (Figure 2C). A finely reticulated pattern of type V collagen birefringence is particularly evident in control tendons (Figure 2E).
 and posterior tibial tendon dysfunction (PTTD) patients (panels B, D, F, G and H) labeled with fluorescein and observed under a fluorescent microscope. Control tendons show parallel orientation of type I (A) and III (C) collagen and strong green fluorescence and a finely reticulated type V collagen (E) network in the interstitium and basement membranes of vessels, coincident with the maintenance of the architecture of the tendons. In contrast, PTTD shows distorted architecture and a diffuse increase of birefringence for all three types of collagen. Type I collagen birefringence is discrete and diffusely distributed in pathologic tendons (B). Type III has the same distribution found for type I but is more prominent and thus defines the fibrosis process installation (D). Type V collagen was mostly found in the vessel walls and around them, resulting in a finely reticulated network (F) when compared to type III fibers. In panels G and H, the strong birefringence of type III and V collagen in the periadvential areas with more vascular density are shown Original magnification X 100 in panels B, D and F; X200 in panels A, C, E, G and H)")
Type I, III and V collagenous fibers in tendon controls (panels A, C and E) and posterior tibial tendon dysfunction (PTTD) patients (panels B, D, F, G and H) labeled with fluorescein and observed under a fluorescent microscope. Control tendons show parallel orientation of type I (A) and III (C) collagen and strong green fluorescence and a finely reticulated type V collagen (E) network in the interstitium and basement membranes of vessels, coincident with the maintenance of the architecture of the tendons. In contrast, PTTD shows distorted architecture and a diffuse increase of birefringence for all three types of collagen. Type I collagen birefringence is discrete and diffusely distributed in pathologic tendons (B). Type III has the same distribution found for type I but is more prominent and thus defines the fibrosis process installation (D). Type V collagen was mostly found in the vessel walls and around them, resulting in a finely reticulated network (F) when compared to type III fibers. In panels G and H, the strong birefringence of type III and V collagen in the periadvential areas with more vascular density are shown Original magnification X 100 in panels B, D and F; X200 in panels A, C, E, G and H)
In contrast, tendon specimens from patients with PTTD show distortion of the architecture and diffuse increase of birefringence for all three types of collagen. Type I collagen birefringence is discrete and diffusely distributed in pathologic tendons (Figure 2B). Type III has the same distribution found for type I (Figure 2B) but is more prominent and thus defines fibrosis process installation (Figure 2D). The distribution pattern of type V collagen was mostly found in and around the vessel walls (Figure 2F), resulting in a finely reticulated network (Figure 2F) when compared to type III fibers (Figure 2D). In Figure 2G, the strong birefringence of type III collagen in the periadvential areas with more vascular density is shown. This observation contrasts with the accentuated vessel wall birefringence in type V collagen, which is found only in areas of incipient angiogenesis (Figure 2H), for which the disease progression results in a pattern similar to that visualized for type III (Figure 2-panel D).
DISCUSSIONPosterior tibial tendon dysfunction (PTTD) is the main cause of acquired flat foot deformity. It is found mainly in adult women and causes pain, longitudinal arch collapse and secondary deformity. Despite its high prevalence worldwide, the exact etiology PTTD has not been conclusively determined.1 Epidemiological studies have shown that trauma does not play a significant role in tendon rupture; however, ageing, obesity and systemic hypertension seem to be associated with PTTD.3 It has also been proposed that tendinopathy is related to changes in matrix turnover, including modifications of collagen synthesis, which could interfere with the physical and structural properties of normal tendons.
Mosier and colleagues have demonstrated a degenerative tendinosis characterized by the absence of inflammatory cells, excessive mucin deposition, fibroblast hypercellularity and neovascularization, which results in marked disruption of collagen bundle structure and fiber orientation.4,5 The collagen fiber structure changes could result in diminished tension strength and spontaneous tendon rupture.
Other studies suggest that a vascular abnormality is involved in the etiology of the syndrome.9 The existence of a critical zone of ischemia that is posterior and distal to the medial malleolus, could predispose the rupture of the posterior tibial tendon and limit its healing process.10,11
Recently, a new etiological theory was proposed in which collagen would have a critical role in the PTTD by promoting changes in the extracellular matrix composition.6
Collagen type I constitutes 95% and collagen types III, IV and V around of 5% of total collagen in normal tendons.12 Collagen type I is responsible for building thick fibers that give resistance to this tissue, while collagen type IV is the main component of basal membrane, and collagen types III and V originate from thin fibrils that intertwine with collagen type I and are responsible for the elasticity of the tendons.
The role of collagen in the tendon has been studied extensively, and rotator cuff injury is one of the best examples of a tendon rupture syndrome related to abnormal collagen synthesis.13,14 It was admitted that tendon degeneration could be caused by repetitive microinjuries, which are subsequently repaired by removal of damaged matrix and deposition of newly abnormal synthesized collagen liable to rupture.13,14
Another example where extracellular matrix remodeling is responsible for tendinopathy was described by Liu et al. (1997)15, who studied the temporal modifications of collagen types I, II and III during the early tendon to bone healing process in their experimental model. They reported an increase in type I, II and III synthesis, albeit only relatively small amounts of type I collagen two weeks after surgery. In addition, type III collagen, resembling Sharpey’s fibers, spanned this interface. This shows a dynamic process involving new synthesis of collagen and modification of the extracellular matrix composition.
More specifically in posterior tibial tendon dysfunction, Gonçalves et al.6 showed through biochemical study an increase of 53.6% in type III collagen and 26.4% of type V collagen and a decrease of 40.4% in the alfa-1 chain and 42.5% in the alfa-2 chain of type I collagen without modification of the total collagen amount. The authors concluded that this different collagen expression pattern found in PTTD was a possible explanation for abnormal fiber synthesis, decreased resistance to mechanical injuries and a predisposition to tendon ruptures.
In addition, Teodoro et al. (2004)8 have reported that type V collagen can be synthesized mainly by smooth muscular cells from blood vessels. The association between type V collagen and blood vessels also interferes epithelial cell migration during the healing process. In the same report the authors also described an increase in type V collagen in neovascularization and tissue remodeling, similar as occurred in our study in PTTD, where the type V collagen was found distributed all over the tendon, but mainly in the perivascular region.
Tendon structure and strength depend on its collagen type composition. Fibrilar types I, II and III collagen maintain tissue architecture and rigidity16, while type V and XI proteins regulate the diameter of collagen fibrils.17–21
As previously described6, the present study shows diminution of type I collagen expression and increased synthesis of types III and V fibrils in PTTD. Comparatively, immunofluorescence analysis of pathologic tendons confirms biochemical findings and demonstrates that collagen remodeling occurs throughout the pathologic tendon. The inflammatory process is negligible in diseased tendons, where type I collagen is found in minor proportion, diffusely distributed, and grossly surrounded by type III fibrils. Type V collagen is augmented in the tendon interstitium but is specifically distributed close to newly formed vessels.
Some aspects deserve comments in our study. First of all is our patients describing tendon obtained after panarthrodesis. These tendons were previously compared to apparently normal tendon obtained in necropsies of patients who died from non-articular causes. The comparison between these tendons and those obtained after panarthrodesis confirmed the normal spatial arrangement of collagen fibers in both groups. The second point relates to the living donor patients in our series, who were also evaluated by the orthopedist surgeon who discounted other causes of tendon changes, such as pain or tendon contracture. The third point that we should emphasize is that all obtained tendons in our study were submitted to collagen evaluation by immunofluorescence, including the macroscopic normal segments. According to this procedure and our results, we postulate that collagen distribution is different along the tibial tendon even though the collagen content may be preserved.
Angiogenesis was a prominent finding observed in our study. The formation of new capillaries in pathologic tendons of patients with PTTD depends on stimulatory and inhibitory proteins that act on endothelial cell receptors. Recently, the influence of endostatin, a potent angiogenesis inhibitor, was described in tendons.22 According to this study, development and maintenance of avascular zones in healthy tendons might have influence over mechanical factors because endostatin concentrations in the supernatants of fibroblast cultures are increased under the influence of intermittent hydrostatic pressure. However, changes in physical tendon properties due to rupture or dysfunction could accelerate the angiogenesis process. Immunostaining of type V collagen in the present study follows the normal pattern of distribution of this fibril in tissues and is normally present in basement membranes or composes heterotypic fibrils with types I and III collagens.
All of these considerations suggest that PTTD should be a final consequence of matrix adaptive remodeling. Predisposing mechanical or ischemic factors could modify tendon collagen composition, characterized by both increased amounts of types III and V fibrils and diminished synthesis of type I collagen, which leads to abnormal fibrillogenesis and production of less resistant fibers that are more susceptible to rupture. Furthermore, tendon matrix remodeling in PTTD is followed by prominent vascular proliferation, possibly in response to an angiogenic stimulus generated by local mechanical changes.