



This study was designed to evaluate the effects of terlipressin versus fluid resuscitation with normal saline, hypertonic saline or hypertonic-hyperoncotic hydroxyethyl starch, on hemodynamics, metabolics, blood loss and short-term survival in hemorrhagic shock.
METHOD:Twenty-nine pigs were subjected to severe liver injury and treated 30 min later with either: (1) 2 mg terlipressin in a bolus, (2) placebo-treated controls, (3) 4 mL/kg 7.5% hypertonic NaCl, (4) 4 mL/kg 7.2% hypertonic-hyperoncotic hydroxyethyl starch 200/0.5, or (5) normal saline at three times lost blood volume.
RESULTS:The overall mortality rate was 69%. Blood loss was significantly higher in the hypertonic-hyperoncotic hydroxyethyl starch and normal saline groups than in the terlipressin, hypertonic NaCl and placebo-treated controls groups (p<0.005). Hyperkalemia (K>5 mmol/L) before any treatment occurred in 66% of the patients (80% among non-survivors vs. 22% among survivors, p=0.019). Post-resuscitation hyperkalemia occurred in 86.66% of non-survivors vs. 0% of survivors (p<0.001). Hyperkalemia was the first sign of an unsuccessful outcome for the usual resuscitative procedure and was not related to arterial acidemia. Successfully resuscitated animals showed a significant decrease in serum potassium levels relative to the baseline value.
CONCLUSION:Hyperkalemia accompanies hemorrhagic shock and, in addition to providing an early sign of the acute ischemic insult severity, may be responsible for cardiac arrest related to hemorrhagic shock.
Resuscitation of patients in hemorrhagic shock remains one of the most challenging aspects of emergency care. Uncontrolled bleeding and subsequent circulatory collapse account for 40 to 45% of trauma-related deaths. Patients with hypovolemia as the primary cause of cardiac arrest rarely survive despite heroic resuscitative measures in the field, with survival rates of 0 to 3.7%.1
An optimal hemorrhagic shock resuscitation strategy during uncontrolled bleeding remains to be determined and should be centered around avoidance of cardiac standstill, which is considered the primary goal of trauma care.
Although current models for hemorrhagic shock research focus on investigation of the pathophysiology of hemorrhage and assessment of therapies, the event cascade involved in hemorrhagic shock decompensation and cardiac arrest has not been fully elucidated.
Hemorrhagic shock is likely to result in electrolyte disorders that may play a role in hemorrhagic shock-related cardiac standstill. Hemorrhagic shock patients are exposed to physiologic processes and life-saving interventions that predispose them to hyperkalemia. Despite hyperkalemia is recognized as a potentially lethal clinical condition, there are few studies addressing hyperkalemia in hemorrhagic shock. One animal study found that elevated serum potassium during prolonged hemorrhagic hypotension was correlated with early death.2 Three studies indicate that an increase in serum potassium occurs during cardiac arrest and prolonged resuscitation.3–5 In the only study assessing hyperkalemia during non-crush trauma resuscitation, the prevalence of hyperkalemia was 29% and all patients who met the criteria for hyperkalemia died.6 In this study, we sought to evaluate the effects of different resuscitation strategies on the hemodynamics, blood loss and metabolic behavior of hemorrhagic shock. Since hyperkalemia was frequently observed, we studied the role of potassium in this uncontrolled bleeding model.
METHODSThe experiments described here were approved by the Institutional Research Ethics Committee and performed in 29 healthy, 3–4 month old male swine (Agroceres® domestic pigs) weighing 27–33 Kg, as a model of hemorrhagic shock (HS) after liver trauma. The animals fasted overnight, but had free access to water. According to current legal requirements and guidelines, animal care was supervised by professional veterinarians.
Animals were premedicated with midazolan (0.4 mg/Kg, im) and ketamine (4 mg/Kg, im). Anesthesia was induced with propofol (1–2 mg/Kg, iv) and was maintained with propofol and fentanil (0.3 mg, and 0.1 μg/Kg/min, respectively, iv). Following oral tracheal intubation, the pigs were mechanically ventilated at a volume-controlled setting of 15 mL/Kg (Servo-Siemens 900C, Germany) with 35% O2. Respiratory frequency was adjusted to 10–15 breaths/min so as to maintain normocapnia. A pulmonary artery catheter was placed by incision into the right internal jugular vein and the left femoral artery was cannulated for mean arterial pressure monitoring and blood sampling. Body core temperature was maintained between 38–39°C throughout the experiment by means of a heating blanket and warmed fluids.
Blood pressure was measured using TruWave transducers (Edwards Lifesciences, Irvine, USA). ECG and arterial pressure were monitored on a Hewlett Packard Merlin Multi-Parameter monitor. Blood was continuously removed from the abdominal cavity using a suction device to determine the exact amount of bleeding.
This model was structured in two phases: the hemorrhagic shock phase and the interventional phase. The Hemorrhagic Shock Phase was begun by means of an incision in the right liver lobe (width, 12 cm; depth 2 cm) after anesthesia, monitoring and laparotomy. The interventional phase was begun when the mean arterial pressure dropped to 40 mmHg with a blood loss of more than 700 mL. For the Interventional Phase, pigs were randomly assigned to five groups: (1) the Terli Group (n=6) received an IV bolus of 2 mg terlipressin; (2) the Control Group (n=5) received an equal volume of saline placebo; (3) the HSS Group (n=6) received 4 mL/Kg of NaCl 7.5%; (4) the HHES Group (n=6) received 4 mL/Kg of NaCl 7.2%/HES 200/0.5 (hypertonic-hyperoncotic hydroxyethyl starch); (5) the NS Group (n=6) received normal saline equivalent to a threefold volume of lost blood. No other resuscitation therapy was administered. At the end of the interventional phase, the surviving animals were followed under another research protocol. Animals with a hemorrhagic shock phase time shorter than 10 minutes were excluded from the study.
Arterial blood (1 mL; ABL555; Radiometer, Copenhagen, Denmark) was sampled and immediately analyzed for oxygen saturation, PaO2, PaCO2, potassium, lactate, bicarbonate, hemoglobin (Hb), pH, and standard base excess. The variables were analyzed at baseline, at the end of the shock phase (before any intervention) and at the end of the interventional phase.
Statistical analysis was performed using SigmaStat software v3.5 (Systat, USA). Data were compared between survivors and non-survivors by means of two-way repeated measures analysis of variance, followed by Student-Newmann-Keuls post-hoc test when indicated. Data was expressed as mean ± SD. P value less than 0.05 was considered as significant.
RESULTSThe survival rate was zero out of five for the Control Group and for the HHES Group, two out of six for the NS Group, three out of six for the HSS Group, and four out of six for the Terli Group.
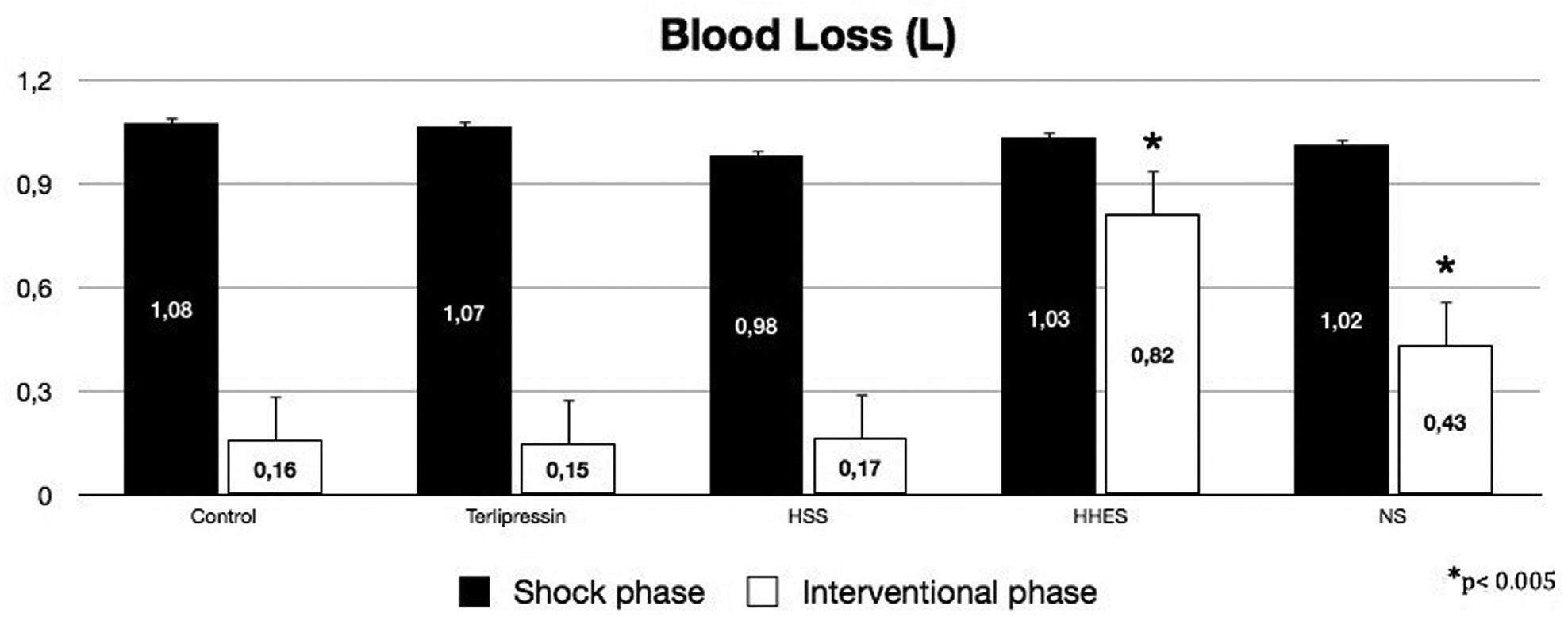
Before liver injury, there were no differences between the hemodynamic and blood variables among groups. The mean overall time of the hemorrhagic shock phase was 20.9 ± 3.9 min and was not significantly different between groups, or between survivors and non-survivors. At the end of the hemorrhagic shock phase, the blood loss and hemodynamic variables were similar for the five groups. At the end of the interventional phase, the intergroup analysis showed similar blood loss for the Terli, HSS and Control Groups, and significantly more blood loss in the HHES and NS groups (Figure 1, p <0.005).
 and at interventional phase (open bars). HSS, hypertonic saline solution; HHES hypertonic-hyperoncotic hydroxyethyl starch; NS, normal saline. *p<0.005 when compared to Control group.")
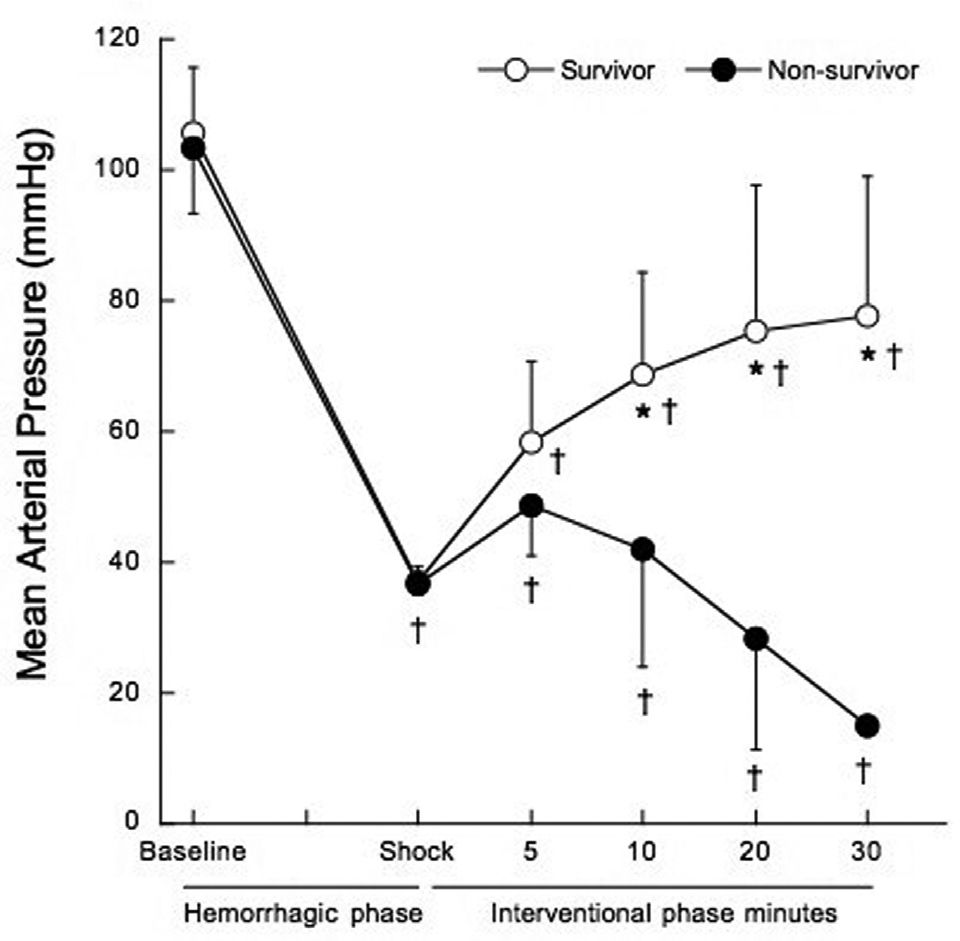
Mean arterial pressure (MAP) five minutes after the shock phase was similar in non-survivors and survivors (48.7±7.6 mmHg vs. 58.2±12.4 mmHg, p=0.060). At 10, 20 and 30 minutes into the interventional phase, MAP progressively decreased in non-survivors, as shown in Figure 2 (p<0.001).
, and after treatment at 5, 10, 20 and 30 minutes of the interventional phase, in survivors (open circles) and non-survivors (filled circles). *p<0.001 when compared to non-survivors. †p<0.05 when compared to baseline.")
Mean arterial pressure before treatment at hemorrhagic phase (baseline and shock), and after treatment at 5, 10, 20 and 30 minutes of the interventional phase, in survivors (open circles) and non-survivors (filled circles). *p<0.001 when compared to non-survivors. †p<0.05 when compared to baseline.
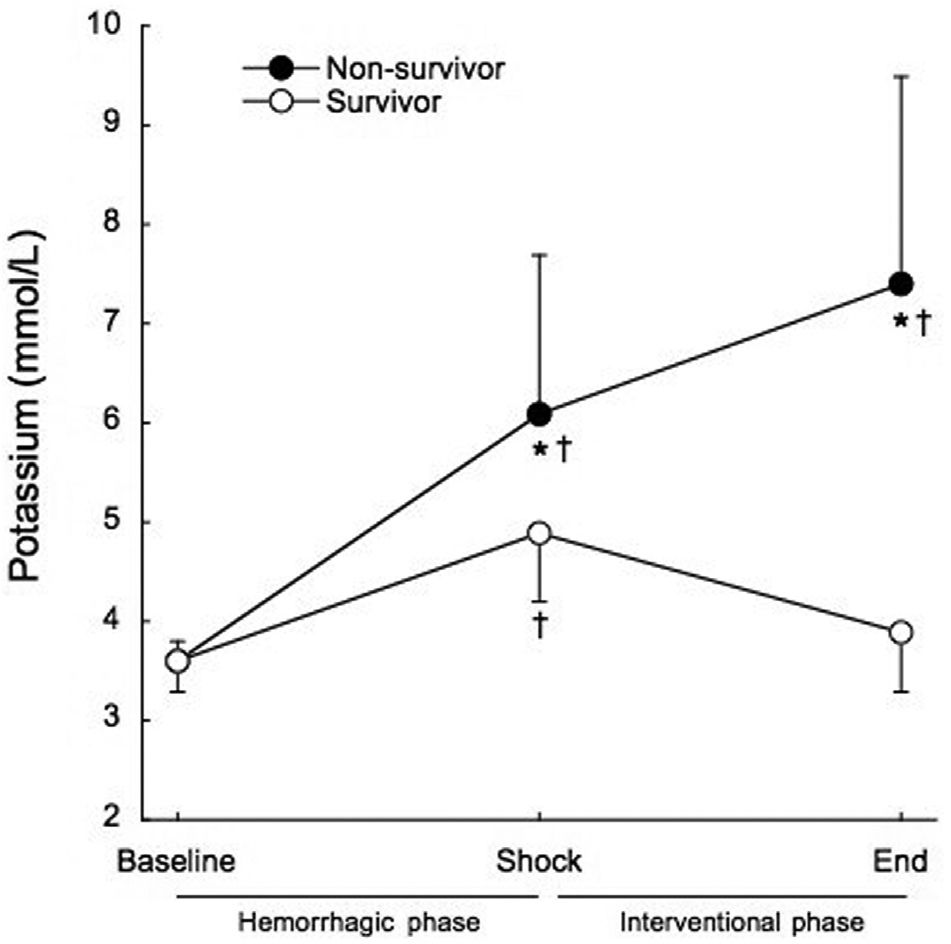
Serum potassium was significantly increased at the end of the shock phase in all groups, before the administration of any experimental therapy (3.6±0.3 mmol/L to 5.8±1.4 mmol/L, p<0.001). The animals that died had significantly higher serum potassium levels at the end of the shock phase than the surviving animals (6.1±1.6 mmol/L vs. 4.9±0.7 mmol/L, p=0.017), as shown in Figure 3. All of the animals that died within the interventional phase presented a further significant increase in the serum potassium levels (6.1±1.6 mmol/L to 7.4±2.1 mmol/L, p=0.014) preceding cardiac arrest. At the end of the interventional phase, the surviving animals showed a significant decrease in serum potassium levels (4.9±0.7 mmol/L to 3.9±0.6 mmol/L, p<0.001). The positive predictive value of potassium levels higher than 5 mmol/L at shock for mortality was 85.7% and the negative predictive value was 70%.
, and after treatment at the end of the interventional phase, in survivors (open circles) and non-survivors (filled circles). †p<0.001 when compared to baseline. *p<0.05 when compared to survivors.")
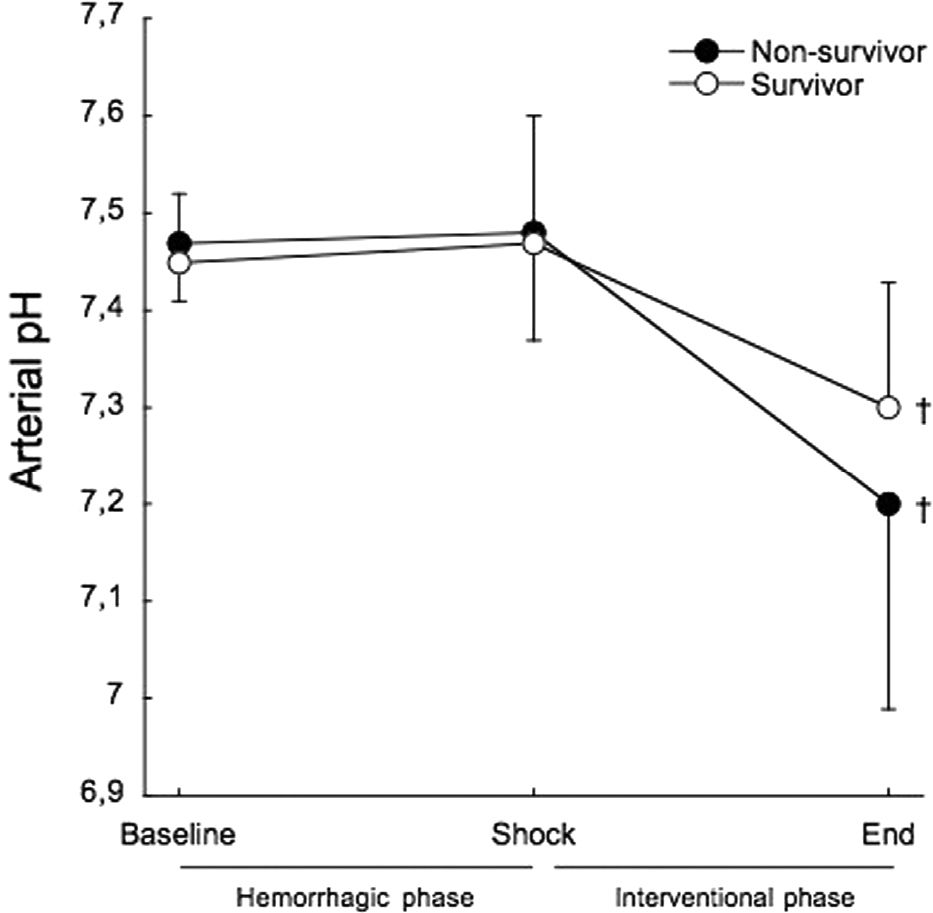
Arterial pH showed no differences between survivors and non-survivors throughout the study (Figure 4). From baseline to shock, there was no variation in arterial pH in both survivors (7.45±0.07 to 7.47±0.13) and non-survivors (7.47±0.06 to 7.48±0.11).
, and after treatment at the end of the interventional phase, in survivors (open circles) and non-survivors (filled circles). †p<0.001 when compared to baseline.")
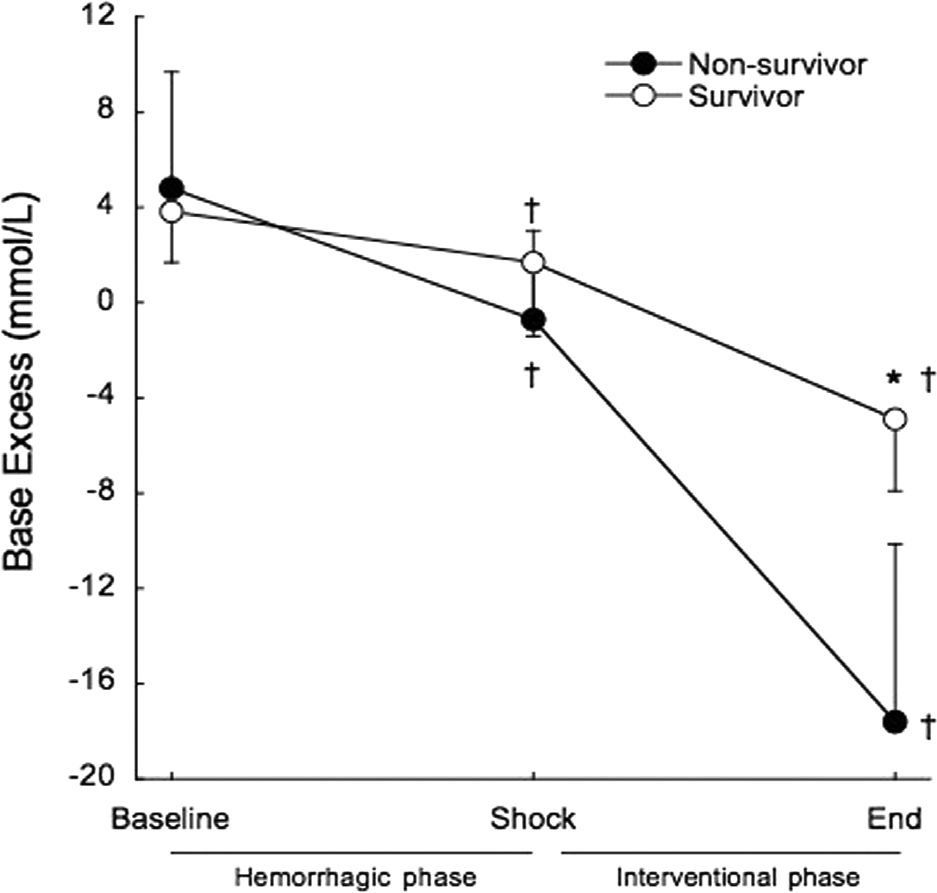
Base excess was significantly decreased at the end of the shock phase in survivors (3.8±2.1 mmol/L to 1.7±3.1 mmol/L, p=0.02) and non-survivors (4.8±7.9 mmol/L to −0.7±3.7 mmol/L, p=0.001), but remained within the normal range and there were no differences between survivors and non-survivors at shock (1.7±3.1 mmol/L vs. −0.7±3.7 mmol/L, p=0.098). At the end of the interventional phase, base excess exhibited a further significant decrease in survivors (1.7±3.1 mmol/L to −4.9±3.0 mmol/L, p<0.001) and non-survivors (−0.7±3.7 mmol/L to −17.6±7.5 mmol/L, p<0.001). Non-survivors presented significantly lower base excess than survivors (p<0.001) (Figure 5).
, and after treatment at the end of the interventional phase, in survivors (open circles) and non-survivors (filled circles). †p<0.01 when compared to baseline. *p<0.001 when compared to non-survivors.")
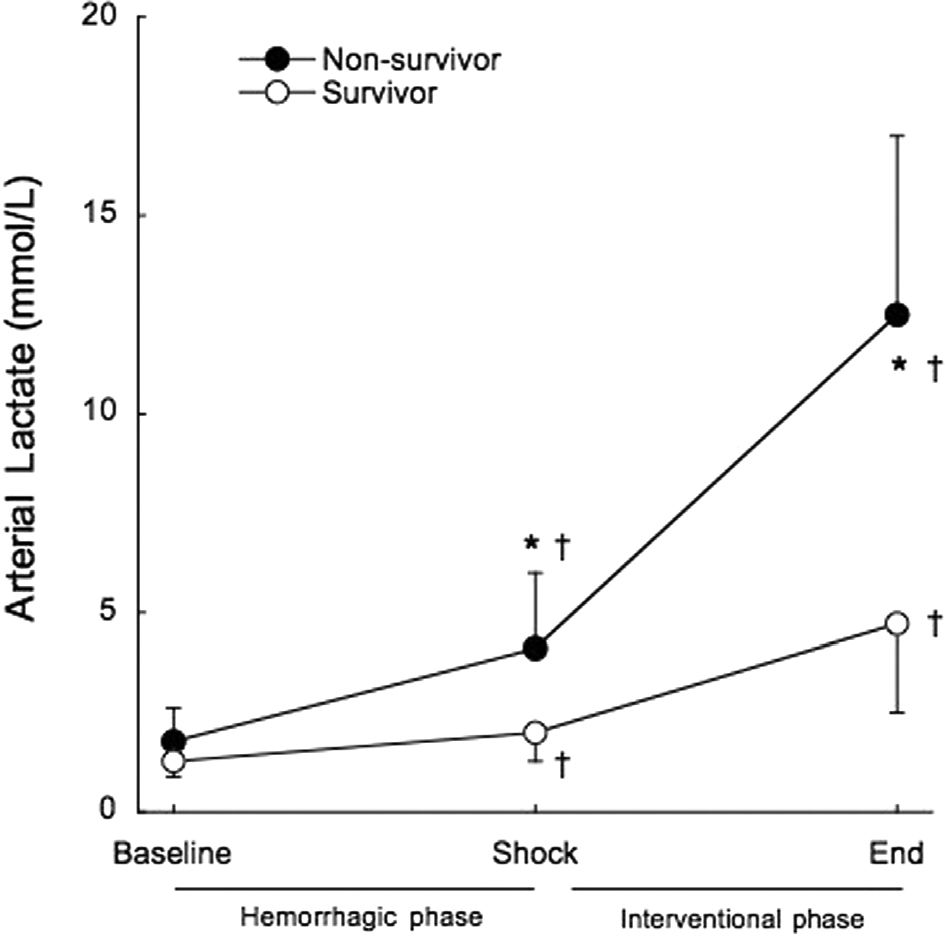
Arterial lactate exhibited a pattern similar to that described for serum potassium, up to a point. It was significantly increased in all groups at the end of the shock phase, before any experimental therapy (1.6±0.6 mmol/L to 3.3±1.7 mmol/L, p<0.001). Non-survivors had significantly higher arterial lactate levels at the end of the shock phase compared to surviving animals (4.1±1.9 mmol/L vs. 2.0±0.7 mmol/L, p=0.002), as shown in Figure 5. All of the animals that died within the interventional phase presented a further significant increase in arterial lactate levels (4.1±1.9 mmol/L to 12.5±4.5 mmol/L, p<0.001) preceding cardiac arrest. However, unlike what was observed for serum potassium, arterial lactate continued to rise significantly in the surviving animals, until the end of the study (2.0±0.7 mmol/L to 4.7±2.2 mmol/L, p=0.014). The positive predictive value of lactate higher than 2 mmol/L at shock for mortality was 62.5% and the negative predictive value was 50% (Figure 6).
DISCUSSION, and after treatment at the end of the interventional phase, in survivors (open circles) and non-survivors (filled circles). †p<0.05 when compared to baseline. *p<0.05 when compared to survivors.")
Main findings of our study were: 1) significant increase in serum potassium occurred before any treatment in all animals; 2) this increase in potassium was not related to arterial acidemia, suggesting rising serum potassium occurs early and accompanies the onset of HS; 3) the rate of rise in serum potassium and its levels before any treatment were higher in non-survivors, suggesting that potassium level is an early sign of acute ischemic insult severity and may early distinguish potential survivors from non-survivors; 4) at the end of study the animals that died presented a further increase in the potassium levels while the surviving animals showed a decrease near to baseline, suggesting that potassium level may be a potential marker of metabolic failure to monitoring the therapeutic strategies in HS.
A significant increase in serum potassium was observed in this animal model of hemorrhagic shock before the administration of any treatment. The mean potassium value at shock was above the upper range of normal. When comparing survivors to non-survivors, the animals that died presented serum potassium levels that were 24.5% higher than non-survivors at shock, before any treatment. This increase in potassium was not related to arterial acidemia. Although a decrease in base excess occurred from baseline to shock, it remained within the normal range and there was no difference between survivors and non-survivors. At five minutes into the interventional phase, mean arterial pressure was unable to differentiate the surviving animals from the non-surviving ones. All of the animals that died presented a further significant increase in the potassium levels preceding cardiac arrest, while the surviving animals showed a significant decrease to near baseline.
Serum lactate levels significantly increased at shock before the administration of any experimental therapy. Although lactate levels were higher in non-survivors at shock, lactate levels presented by non-survivors at shock were similar to those presented by survivors at the end of the experiment. The continued rise in serum lactate presented by survivors is probably the result of the washout of products of anaerobic metabolism during reperfusion. This post-resuscitation lactic acidemia in survivors was accompanied by a decrease in serum potassium.
Hemorrhagic shock is a global ischemic insult. Within seconds of the onset of ischemia, oxidative phosphorylation is inhibited and inefficient anaerobic metabolism becomes the primary means of high energy phosphate production. Once the alternative energy sources are exhausted, the disruption of the energetic status of the cell leads to extracellular K accumulation.7 Interstitial hyperkalemia during hemorrhage has previously been demonstrated.8–10 Three mechanisms are recognized as contributing to disruption of potassium homeostasis during ischemia: 1) increased cellular K efflux, 2) decreased cellular K influx and 3) shrinkage of extracellular space.7,11 Despite experimental evidence suggested that the Na-K pump is inhibited during ischemia and thus is a mechanism contributing to the decrease K influx7,12, the ATP-sensitive K (KATP) channels activation are thought to be the primary mechanism through which K efflux may increase during ischemia and contribute to the rise in potassium.11,13,14 There is an ongoing debate on the literature about the role of KATP channels in hemorrhagic shock decompensation.15–17 However, based on our findings, we can hypothesize that KATP channels activation would occur earlier, by the time of HS onset.
Despite hyperkalemia is recognized as a potentially lethal clinical condition, there are few studies addressing hyperkalemia in hemorrhagic shock. The silent characteristic of hyperkalemia, which may be not recognized in the patient who dies early during resuscitation, as well its transient behavior, in those who undergo a successful resuscitation, may be responsible for the scarce attention to this complication.
Torres et al.2 found that elevated serum potassium during prolonged hemorrhagic hypotension in rats was correlated with mortality and may explain the sudden death in animals in their experiment.
Perkins et al,6 in a prospective observational analysis of 131 patients during the initial 12 hours after admission for non-crush trauma, found a surprisingly high prevalence of hyperkalemia (29%) and all patients who met the criteria for hyperkalemia died. In this original report, independent risk factors for hyperkalemia were plasma potassium level at admission ≥ 4 mmol/L and transfusion of cell- or plasma-based products. Acidosis did not contribute to hyperkalemia.
The most prominent effect of hyperkalemia is on the myocardium. Decreases in resting membrane potential reduce myocardial conduction velocity and increase the rate of polarization. A slowed conduction velocity (increased PR interval and widened QRS complex), especially in the presence of increased repolarization rates (peaked T waves), increases the chance of ventricular fibrillation and cardiac standstill.
In their assessment of hyperkalemia and hypocalcemia during cardiac arrest in an animal model of ventricular fibrillation, Niemann et al.5 found that hyperkalemia and hypocalcemia occurred during prolonged resuscitative efforts in unsuccessfully resuscitated animals. The increase in serum potassium observed in their study, in agreement with our data, was not related to acidemia and successfully resuscitated animals did not demonstrate these electrolyte changes. The authors hypothesized that the electrolyte changes may play a role in refractory cardiac arrest.
Martin et al.3,4 showed that an increase in serum potassium occurs during cardiac arrest and prolonged cardiopulmonary resuscitation in an animal model and in patients.
Rather than being only a sign of acute ischemic insult severity, hyperkalemia is a life-threatening condition that requires emergency treatment, mainly before blood transfusion, which is a cornerstone in the treatment of hemorrhagic shock. Brown et al.18 retrospectively studied 138 cardiac arrests in a pediatric population, comparing patients who received a rapid blood transfusion with those who did not, found that the during cardiac arrest mean serum potassium in the non-transfused group versus the transfused group was 5.6 mmol/L versus 8.2 mmol/L. Data from their study demonstrated that hyperkalemia during cardiac arrest could be explained as a consequence of rapid RBC transfusion during hypovolemic-reduced cardiac output states.
A number of reports describing transfusion-related hyperkalemia and cardiac arrest have been published.19–22 Smith et al.22 in a recent retrospective review, identified 16 patients in whom stored red blood cell (RBC) transfusion-associated hyperkalemia was found to be a significant factor in precipitating intraoperative cardiac arrest. The main finding of the study was that transfusion-associated hyperkalemic cardiac arrest may develop with rapid RBC administration even with modest transfusion volume. Banked RBC potassium concentration may present levels up to 35 mmol/L after 21 storage days.23
The most fundamental limitation of this study is that it is involved in an animal model, and so the way it is related to the complexity of real human injuries is unclear. The study was not addressed to evaluate the serum potassium behavior patterns during hemorrhagic shock and we were not able to analyze serum ionized calcium. The heterogeneity of the tested strategies recommends caution in the analysis of the registered results after the various therapeutic interventions.
If these results could be translated to human beings, they may open a new vista into the resuscitative strategy for hemorrhagic shock. Current animal models used to evaluate different resuscitative strategies focused on hemodynamic data will need an urgent review and our clinical resuscitative protocols will need to incorporate identification and prophylactic management of high-risk patients.
CONCLUSIONTo the best of our knowledge, this is the first demonstration that, during uncontrolled bleeding, hyperkalemia unrelated to arterial acidemia accompanies the onset of hemorrhagic shock and relates to mortality. Rising potassium may be one early sign of acute ischemic insult severity and could conceivably help us clarify the frontier of hemorrhagic shock refractoriness.
Studies are needed to evaluate the mechanisms and the clinical impact of rising potassium in hemorrhagic shock.