





IgG4-related systemic sclerosing disease (IRSSD) is a new clinical entity that is characterized by elevated levels of serum IgG4; plasma cell infiltrates in tissues, including the pancreas, lung, liver, kidney and breast; and a good response to corticosteroid therapy.1–4 We report a case of a patient with sclerosing cholangitis and pulmonary inflammatory pseudotumors. The diagnosis was made twelve years after the onset of the initial symptoms. The aim of this report is to describe a case with different radiological patterns.
CASE REPORTA 70-year-old male patient was referred to our institution in November 2009 for the evaluation of recurrent pulmonary infiltrates. His clinical problems started in 1998. The patient was diagnosed with sclerosing cholangitis after a cholecystectomy for gallstones. Dilatation of the bile ducts using endoscopic retrograde cholangiopancreatography was performed. Symptomatic control was achieved using ursodeoxycholic acid. No inflammatory bowel disease was detected. In addition, the patient has suffered from diabetes since 2000 and became insulin-dependent just after diagnosis. In 2004, a lower lobe pulmonary nodule was detected in a routine abdominal computed tomography (CT). This same exam revealed no signs of liver disease but demonstrated atrophy of the pancreas and irregularities in the pancreatic ducts, suggesting chronic pancreatitis. The pulmonary nodule was resected and diagnosed as an inflammatory pseudotumor. Four years later, the patient developed recurrent episodes of dyspnea and fatigue. Chest CTs over the years showed different image patterns and indicated predominantly bilateral peripheral nodules or subpleural consolidations with a ground-glass halo (Figure 1 and 2). The symptoms and signs improved after treatment with prednisone but reoccurred after tapering off of the corticosteroid treatment. Several tests were performed to look for evidence of autoimmune diseases, but these tests yielded negative results. The association of relatively rare conditions, such as an indolent sclerosing cholangitis, presumed autoimmune pancreatitis with endocrine insufficiency, and inflammatory pseudotumors of the lung, led us to the diagnosis of IRSSD. The serum IgG4 concentration was 936 UI (reference value: <140 UI). An anatomopathological revision of the previous pulmonary nodule biopsy revealed a dense fibrosis with plasmacytic infiltration and focal lymphoid aggregation in the nodular lesion. Small numbers of eosinophils were detected (<5 cells/high-power field [HPF] in most areas). Some vessels were partially or completely obstructed by inflammatory cells. Most of the infiltrating plasma cells were positive for IgG4, and the IgG4/IgG ratio was greater than 90% (Figure 3). Light chain restriction (normal range of kappa:lambda) and Epstein-Bar virus encoded RNA, which was evaluated using in situ hybridization, were not detected. There were no signs of malignancy.
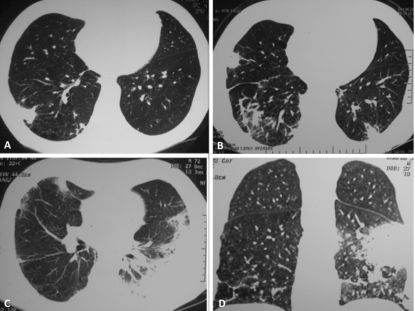
 scans showing different image patterns along the years. A: December, 2004: the initial clinical presentation as a subpleural solitary pulmonary nodule in the right inferior lobe with adjacent subpleural opacities. The nodule was resected and diagnosed as an inflammatory pseudotumor. B: Three years later, CT scans with multiple spiculated pulmonary nodules surronded with ground glass halo and architectural distortion of the adjacent parenchyma. C and D: June, 2008: Pulmonary nodule with ground glass halo in the middle lobe along with consolidation in the left inferior lobe resembling organizing pneumonia. There was a complete response after corticosteroid treatment.")
Chest computer tomography (CT) scans showing different image patterns along the years. A: December, 2004: the initial clinical presentation as a subpleural solitary pulmonary nodule in the right inferior lobe with adjacent subpleural opacities. The nodule was resected and diagnosed as an inflammatory pseudotumor. B: Three years later, CT scans with multiple spiculated pulmonary nodules surronded with ground glass halo and architectural distortion of the adjacent parenchyma. C and D: June, 2008: Pulmonary nodule with ground glass halo in the middle lobe along with consolidation in the left inferior lobe resembling organizing pneumonia. There was a complete response after corticosteroid treatment.

Chest CT scans obtained in 2009. On the left column, solitary pulmonary nodule in the upper right lobe along with parenchimal bands in the inferior right lobe. Five months later, the right upper nodule disappeared, but other nodules surrounded with ground glass halo appeared in the inferior lobes bilaterally.
; B: Plasmacytic infiltration of the fibrous lesion (H&E stain, magnification × 100); C: Occlusive vascular lesion with plasmacytic infiltration (H&E stain, magnification × 100); D: Immunopositivity for IgG4 in the infiltrating plasma cells (Verhoeff-Masson trichrome stain, original magnification × 100).")
Lung biopsy of the pulmonary nodule. A: Nodular fibrous lesion with immunopositivity for IgG4 in the infiltrating plasma cells (Verhoeff-Masson trichrome stain, original magnification x 2); B: Plasmacytic infiltration of the fibrous lesion (H&E stain, magnification × 100); C: Occlusive vascular lesion with plasmacytic infiltration (H&E stain, magnification × 100); D: Immunopositivity for IgG4 in the infiltrating plasma cells (Verhoeff-Masson trichrome stain, original magnification × 100).
The patient received prednisone (20 mg/daily) with recrudescence of the tomographic findings. We prescribed a low dose of prednisone (10 mg/daily) to avoid recrudescence of the pulmonary nodules.
DISCUSSIONHigh serum IgG4 that is associated with sclerosing pancreatitis was first described in 2001 by Hamano and colleagues and suggests a distinct disease entity due to its responsiveness to corticosteroids.5 This report and other reports have indicated the involvement of multiple organs containing inflammatory pseudotumors with plasma cells expressing IgG4, such as the pancreas, bile ducts, gallbladder, breast, salivary glands, retroperitoneum, kidney, lung, and prostate.1–4 Recently, a new clinical entity, IRSSD, has been proposed as an etiology for hyper-IgG4 gammaglobulinemia and IgG4 plasma cell infiltrates in multiple organs and is responsive to glucocorticoids.1–3,6
Zen et al. examined clinical and histological features of inflammatory pseudotumors and described lesions that are characterized by dense lymphoplasmacytic infiltrates intermixed with fibrosis, eosinophilic infiltration, irregular narrowing of the bronchioles that are entrapped in nodules, obliterative phlebitis or arteritis, and an interstitial pneumonia pattern at the boundaries of nodules. Immunostaining revealed many IgG4+ plasma cells diffusely distributed within the nodules, and the ratio of IgG4+ plasma cells to other plasma cells was extraordinarily high.7–8
Convincing clinical criteria have not been clearly defined. IgG4 serum levels are frequently elevated in the majority of patients (above 140 mg/dl). However, 25% of patients display normal IgG4 levels.2 Immunostaining has revealed IgG4+ plasma cells. IgG4+ cells that number between >60 and 100 cells/high-power field (HPF) and a ratio IgG4+/IgG+ cells between >40% and 50% are highly suggestive of IgG4-related disease.9
Although its prevalence is still unknown, previous studies have reported an increased incidence in male patients in their sixties who usually present with dry cough or dyspnea or are asymptomatic.1 Asian patients seem to have a genetic predisposition for IRSSD.10 However, cases in non-Asian patients, including the current case subject, have also been reported.
Due to the heterogeneity of radiological presentations, IRSSD must be included as a differential diagnosis of other interstitial diseases. IRSSD is categorized into four major subtypes: the solid nodular type; the round-shaped ground-glass opacity type; the alveolar interstitial type, which includes honeycombing, bronchiectasis, and diffuse ground-glass opacities; and the bronchovascular type, which is characterized by thickening of the bronchovascular bundles and interlobular septa.11 To our knowledge, we report herein the case with the longest radiological follow-up (seven years) and the first case with multiple documented radiological patterns in the same patient. In addition, we noticed a pattern of consolidation as a possible pulmonary involvement of IRSSD.
Corticosteroids appear to be effective in the majority of the reported cases. The most commonly used dosage is prednisolone at 1 mg/kg/day with gradual tapering every 1-2 weeks for a total treatment period of 11 weeks. Clinical reports have also indicated spontaneous resolution without treatment.10 However, relapses during corticosteroid tapering are also possible, as in this current case. Therefore, low dose prednisone may be necessary as a maintenance treatment. Alternative drug regimens for corticosteroid-resistant cases have not been studied. Treatment data were extrapolated from clinical studies of patients with pancreatic or biliary disease. The drug regimens used for the treatment of lung disease were empirical.
In conclusion, when faced with inflammatory pseudotumors (histologically corresponding to plasma cell granuloma) or migratory pulmonary infiltrates that respond to corticosteroid therapy, the diagnosis of IRSSD should be considered in the differential diagnosis. Accurate diagnoses may lead to effective treatments and prevent unnecessary lung resections.
No potential conflict of interest was reported.
Dias OM was responsible for the patient's diagnosis, wrote the first draft of the manuscript, conceived the review of the literature on the subject. Kawassaki AM was involved in the review and critical revision of manuscript before submission. Cukier A was involved in the clinical care of the patient, also participated in the manuscript review and critical revision before submission. Haga H made the anatomopathological review and performed the immohistochemistry analysis of the lung biopsy. Carvalho CRR was involved in the manuscript review and critical revision before submission. All authors have read and approved the final manuscript.