




The objective of this study was to evaluate the involvement of peripheral nitric oxide (NO) in vagotomy-induced pulmonary edema by verifying whether the nitric oxide synthases (NOS), constitutive (cNOS) and inducible (iNOS), participate in this mechanism.
INTRODUCTION:It has been proposed that vagotomy induces neurogenic pulmonary edema or intensifies the edema of other etiologies.
METHODS:Control and vagotomized rats were pretreated with 0.3 mg/kg, 3.0 mg/kg or 39.0 mg/kg of L-NAME, or with 5.0 mg/kg, 10.0 mg/kg or 20.0 mg/kg of aminoguanidine. All animals were observed for 120 minutes. After the animals' death, the trachea was catheterized in order to observe tracheal fluid and to classify the severity of pulmonary edema. The lungs were removed and weighed to evaluate pulmonary weight gain and edema index.
RESULTS:Vagotomy promoted pulmonary edema as edema was significantly higher than in the control. This effect was modified by treatment with L-NAME. The highest dose, 39.0 mg/kg, reduced the edema and prolonged the survival of the animals, while at the lowest dose, 0.3 mg/kg, the edema and reduced survival rates were maintained. Aminoguanidine, regardless of the dose inhibited the development of the edema. Its effect was similar to that observed when the highest dose of L-NAME was administered. It may be that the non-selective blockade of cNOS by the highest dose of L-NAME also inhibited the iNOS pathway.
CONCLUSION:Our data suggest that iNOS could be directly involved in pulmonary edema induced by vagotomy and cNOS appears to participate as a protector mechanism.
Neurogenic pulmonary edema (NPE) is a fatal complication of severe insults to the central nervous system.1,2 It is proposed that neurogenic pulmonary edema is a functional disturbance provoked by adverse stimuli from outside the lungs and that in the rat, the pulmonary afferent fiber is essential to the production of this edema.3 Autonomic nervous dysfunction, possibly sympathetic nerve over-excitation or vagus nerve dysfunction by vagotomy or by lesion of vagal nuclei in the medulla, has been show to induce NPE.2 Studies involving NPE by intracisternal injection of fibrinogen and thrombin have indicated that bilateral vagotomy or the treatment with atropine increased the severity of the edema.4 Vagotomy increases the amount of edema for a given degree of pulmonary hypertension.5 Vagotomy-induced pulmonary edema has been discussed as neurogenic pulmonary edema. The lungs of vagotomized rats showed alveolar edema.6 Blood volume and hematocrit reading became considerably reduced during the development of acute pulmonary edema caused by bilateral cervical vagotomy, depending on whether pulmonary hemorrhage occurs as a complication of the edema and congestion.7
Since 1966, it has been proposed that bilateral interruption of afferent impulses of the tenth cranial nerve is the factor that initiates vagotomy-induced lung edema.8 A subsequent observation concluded that vagal capsaicin-sensitive nerves wielded an inhibitory effect on the development of fibrin injection into the cisterna magna-induced pulmonary edema.9 It was proposed that an unknown neurotransmitter released from capsaicin-sensitive nerves may participate in increasing the lung vascular permeability caused by sympathetic nerve stimulation and that norepinephrine may also play a role in the regulation of permeability through alpha- and beta-adrenoceptors.10
It has been recognized that the main site of nitric oxide (NO) production in the circulatory system is in the lungs.11 NO is produced by a group of enzymes known as nitric oxide synthases (NOS). These enzymes convert L-arginine into NO and L-citruline. Three isoforms of NOS have been identified, including two constitutive forms: neuronal (nNOS) and endothelial (eNOS), and an inducible form (iNOS). NO is a potent vasodilator in bronchial circulation and may play an important role in regulating airway blood flow. It also modulates vascular tone through its vasodilatory properties. Excess amounts of NO may cause hypotension associated with sepsis, and decreased NO levels within the lungs may contribute to the pathologic states associated with pulmonary hypertension. NO may also play a critical role in ventilation-perfusion coupling in the lung. This theory is supported by the fact that endogenous NO levels in the lung change rapidly in direct proportion to inspired oxygen.12
NO has been related to pulmonary edema of various etiologies. Pulmonary exhaled NO was lower in mountaineers prone to high-altitude pulmonary edema than in those resistant to this condition.13,14 Reduced exhaled NO may be as a result of altered pulmonary NO synthesis and/or transport and clearance, in line with the hypothesis that, in these subjects, a defect in pulmonary epithelial NO synthesis may contribute to exaggerated hypoxic pulmonary vasoconstriction and, in turn, to pulmonary edema.13 It was also observed that susceptible subjects have decreased nitrate-nitrite concentrations in bronchoalveolar fluid at high altitude, while resistant subjects have increased concentrations, further supporting a critical role for endogenous NO production in maintaining lower pulmonary vascular resistance.15 L-Arginine, a NO synthase substrate, and N-nitro-L-arginine (L-NNA), a NO synthase inhibitor, prevented and aggravated, respectively, the increase in pulmonary vascular permeability induced by radiologic contrast medium at high doses, in rats.16 Pretreatment with NOS inhibitors, such as N-nitro-L-arginine methyl ester (L-NAME), aminoguanidine and dexamethasone, significantly reduced endotoxin-induced pulmonary edema. Overproduction of NO was considered detrimental to the lung and exerted toxic effects on the pulmonary endothelial layer.11 However, the involvement of NO in NPE has not yet been evaluated.
The main objective of this study was to evaluate the involvement of peripheric NO and its enzymatic production in rats with vagotomy-induced pulmonary edema, by assessing the participation of the cNOS and iNOS pathways in this mechanism. To this end, some animals were pretreated with L-NAME, a cNOS inhibitor, and others with aminoguanidine, a relatively specific iNOS inhibitor.
MATERIALS AND METHODSAnimals and experimental groupsExperimental protocols were performed in accordance with the Guide for the Care and Use of Laboratory Animals and the Ethical Principles for Animal Experimentation, established by the Brazilian Committee for Animal Experimentation (COBEA), and in accordance with the animal experimentation Ethics Committee of the State University of Londrina (CEEA/UEL).
Adult male Wistar rats (n = 167), weighing 250–320 g, were distributed into 14 groups: C—control animals (n = 14); V—vagotomized animals (n = 14); CL 0.3 (n = 11); CL 3.0 (n = 11); CL 39 (n = 11)—control animals pretreated with 0.3 mg/kg, 3.0 mg/kg or 39.0 mg/kg L-NAME, respectively; VL 0.3 (n = 14); VL 3.0 (n = 9); VL 39 (n = 8)—vagotomized animals pretreated with 0.3 mg/kg, 3.0 mg/kg or 39.0 mg/kg L-NAME, respectively; CA 5 (n = 11); CA 10 (n = 11); CA 20 (n = 11)—control animals pretreated with 5.0 mg/kg, 10.0 mg/kg or 20.0 mg/kg aminoguanidine, respectively; VA 5 (n = 14); VA 10 (n = 14); VA 20 (n = 14)—vagotomized animals pretreated with 5.0 mg/kg, 10.0 mg/kg or 20.0 mg/kg aminoguanidine, respectively.
Treatment with nitric oxide synthase (NOS) inhibitorsPretreatment consisted of an intravenous injection of L-NAME or aminoguanidine 10 minutes before surgery. Untreated control and vagotomized animals received an equivalent volume of saline. All animals were anesthetized by inhalation of ethyl ether.
The drugs utilized in this study were: N(G)-nitro-L-arginine methyl ester, L-NAME (RBI) and aminoguanidine (Sigma Co., St Louis, MO, USA).
VagotomyIn controls and vagotomized rats, the vagus nerve was exposed through a midline incision in the neck. The vagus nerve was identified and separated from the common carotid artery. In vagotomized animals, the vagus nerve was severed bilaterally. After recovering from surgery, all animals were observed for 120 minutes. Those that died during this period were noted, along with their time of death. Survivors were sacrificed, under ether anesthesia by severing the inferior vena cava and draining the blood.
After the animals' death, the trachea was cannulated in order to observe tracheal fluid and to classify the severity of pulmonary edema. By way of thoracic incision, lungs were removed and weighed to evaluate pulmonary weight gain and edema index.
Pulmonary edema evaluationThe severity of pulmonary edema was graded by a method proposed by Hamdy et al., with modifications.17 The edema was graded from 0 to 2, according to the presence of fluid in the trachea, with 0 for none and 2 for severe. When edema fluid spontaneously appeared in the tracheal tube after opening the chest, the edema was classified as grade 2. If fluid appeared in the tracheal tube only when the lungs were gently pressed, the edema was grade 1. When edema fluid did not appear at all, even under gentle pressure, the grade was 0.
The lungs were weighed to obtain the weight gain caused by pulmonary edema. As previously proposed, edema was evaluated from the ratio of pulmonary weight:body weight (100xPW/BW); this ratio is still in use as the edema index.18
Statistical analysisDifferences between means and the SE were examined for significance by ANOVA, followed by Tukeýs Multiple Comparison Test. Survival time was examined by Student's t-test, comparing each pair of control to vagotomized animals, vagotomized to vagotomized pretreated with drugs and the pairs of control and vagotomized animals pretreated with drugs. Survival time was displayed in curves.
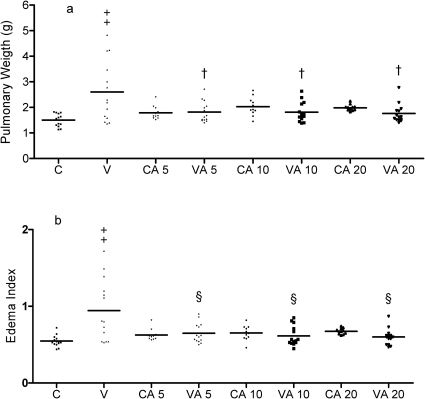
RESULTSOur data showed that vagotomy promoted the development of pulmonary edema, as, in the vagotomized rats, the pulmonary weight (g) (2.60±0.31) and edema index (0.94±0.10) were higher than the pulmonary weight (g) (1.50±0.06) and edema index (0.54±0.01) of the control animals (Figures 1 and 2). Among vagotomized rats (n = 14), four animals died during the experimental period (Figure 3) and five animals developed grade 2 edema (Tables 1 and 2).
 pulmonary weight (g) and (B) edema index. Controls or vagotomized rats pretreated with saline (C and V) or with 0.3 mg/kg (CL 0.3 and VL 0.3), 3.0 mg/kg (CL 3.0 and VL3.0), or 39 mg/kg (CL 39 and VL 39) L-NAME. * Statistically different from its pair control (p<0.01), † statistically different from vagotomized (p<0.05), ‡ statistically different from VL 39 (p<0.01).")
Mean and individual values of (A) pulmonary weight (g) and (B) edema index. Controls or vagotomized rats pretreated with saline (C and V) or with 0.3 mg/kg (CL 0.3 and VL 0.3), 3.0 mg/kg (CL 3.0 and VL3.0), or 39 mg/kg (CL 39 and VL 39) L-NAME. * Statistically different from its pair control (p<0.01), † statistically different from vagotomized (p<0.05), ‡ statistically different from VL 39 (p<0.01).
 pulmonary weight (g) and (B) edema index. Controls or vagotomized rats pretreated with saline (C and V) or with 5 mg/kg (CA 5 and VA 5), 10 mg/kg (CA 10 and VA 10) or 20 mg/kg (CA 20 and VA 20) aminoguanidine. † statistically different from vagotomized (p<0.01), ‡statistically different from control group (p<0.001), § statistically different from vagotomized (p<0.001).")
Mean and individual values of (A) pulmonary weight (g) and (B) edema index. Controls or vagotomized rats pretreated with saline (C and V) or with 5 mg/kg (CA 5 and VA 5), 10 mg/kg (CA 10 and VA 10) or 20 mg/kg (CA 20 and VA 20) aminoguanidine. † statistically different from vagotomized (p<0.01), ‡statistically different from control group (p<0.001), § statistically different from vagotomized (p<0.001).
 Survival curve of controls (C), vagotomized rats (V) or rats pretreated with 0.3 mg/kg (CL 0.3and VL 0.3), 3.0 mg/kg (CL 3.0 andVL3.0) or 39 mg/kg (CL39 and VL 39) L-NAME . (B) Survival curve of controls (C), vagotomized rats (V) or rats pretreated with 5 mg/kg (CA 5 and VA 5), 10 mg/kg (CA 10 and VA 10) or 20 mg/kg (CA 20 and VA 20) aminoguanidine. The upper horizontal line represents 100% survival during the period of observation (120 minutes). On the other lines, each drop is equivalent to the death of one animal. *Statistically different from control group (p<0.05), † statistically different from VL 39 (p<0.05).")
(A) Survival curve of controls (C), vagotomized rats (V) or rats pretreated with 0.3 mg/kg (CL 0.3and VL 0.3), 3.0 mg/kg (CL 3.0 andVL3.0) or 39 mg/kg (CL39 and VL 39) L-NAME . (B) Survival curve of controls (C), vagotomized rats (V) or rats pretreated with 5 mg/kg (CA 5 and VA 5), 10 mg/kg (CA 10 and VA 10) or 20 mg/kg (CA 20 and VA 20) aminoguanidine. The upper horizontal line represents 100% survival during the period of observation (120 minutes). On the other lines, each drop is equivalent to the death of one animal. *Statistically different from control group (p<0.05), † statistically different from VL 39 (p<0.05).
Incidence of edema grade of controls (C), vagotomized (V) animals and controls or vagotomized animals pretreated with 39 mg/kg (CL 39 and VL 39), 3.0 mg/kg (CL 3.0 andVL3.0) or 0.3 mg/kg (Cl 0.3 and VL 0.3) of L-NAME.
Incidence of edema grade of controls (C), vagotomized (V) animals and controls or vagotomized animals pretreated with 20 mg/kg (CA 20 and VA 20), 10 mg/kg (CA 10 and VA 10) or 5 mg/kg (CA 5 and VA 5) of aminoguanidine.
The effects observed in vagotomized animals pretreated with L-NAME varied according to the dose (Figure 1). The highest dose, 39 mg/kg, reduced the edema index (0.58±0.02) and no animal in the group VL39 suffered edema grade 2 (Table 1). On the other hand, at the lowest dose, 0.3 mg/kg, the pulmonary weight (g) (2.97±0.28) and edema index (1.00±0.10) remained high and, out of the 14 animals in the group VL0.3, 6 had edema grade 2 (Table 1). The dose of 3.0 mg/kg promoted intermediate effects between the other two doses (Figure 1). Treatment with 39 mg/kg also prolonged the survival of the animals while, in the group pretreated with 0.3 mg/kg L-NAME, eight rats died during the observation period (Figure 3A).
The results observed in control animals treated with L-NAME or with aminoguanidine were similar to those in the control animals without treatment. All three doses of aminoguanidine treatment reduced the pulmonary weight, the edema index and the edema grade in vagotomized rats (Figure 2 and Table 2).
DISCUSSIONThe objective of the present study was to assess the involvement of NO pathways in the development of pulmonary edema. It was observed that the largest dose of L-NAME inhibited the development of pulmonary edema induced by vagotomy in the group VL39 and prolonged the survival of those animals, while the smallest dose did not promote the same effects. On the other hand, treatment with aminoguanidine, regardless of the dose administered, inhibited the development of the edema. Our data suggest that NO production from iNOS is involved in the pulmonary edema induced by vagotomy and, on the contrary, NO derived by cNOS seems to have protective effects against this type of edema.
In rats, the vagus nerve and the sympathetic trunk are anatomically adjacent. Hence, in the vagotomy performed in the present study, the sympathetic branch could be damaged, too. In early studies of pulmonary edema following vagotomy it was shown that when the midcervical vagosympathetic trunk in guinea pigs was severed, bilateral interruption of afferent impulses of the tenth cranial nerve was the factor that initiated lung edema.8 This edema was developed in artificially ventilated guinea pig,19 and tracheotomy had no demonstrable effect in vagotomized rats submitted to moderate pulmonary congestion.7 The observation that edema induced by fibrinogen and thrombin was increased by bilateral vagotomy, or by treatment with atropine, indicates that efferent cholinergic fibers in the vagus nerve may play a role in lowering the severity of pulmonary edema.4 In a subsequent experiment involving the use of capsaicin, it was observed that afferent vagal impulses could exert an inhibitory effect on the development of fibrin-induced NPE.9 Moreover, it was observed that pretreatment with atropine attenuated pulmonary edema in spinal cord-injured rats.20 A hypothesis was introduced that early administration of high atropine doses can prevent NPE development on the basis of the attenuation of heart rate decrease, which was recognized as an important factor of neurogenic pulmonary edema formation.21 In rats, bilateral vagotomy increased the incidence of NPE.17 The rats that suffered vagotomy in our study developed a higher pulmonary edema index than the controls.
Among vagotomized rats, five animals showed edema grade 2, and two had edema grade 1. Similar results were observed in rats treated with a dose of 0.3 mg/kg of L-NAME, which had a high edema index. Although these results represent development of edema, not all animals showed a degree of edema. In our opinion, this is because of the fact that the rats were observed for a period of only 120 minutes and not until death, when more animals would possibly have developed edema. The establishment of an observation period followed by sacrifice of those who survived could also justify the degree of variance observed in the results presented.
The incidence of pulmonary edema in rats promoted by intracisternal injection of fibrinogen and thrombin associated with vagotomy was related to the high (25 mg/kg or 50 mg/kg) doses of pentobarbital anesthesia.4 It was later observed that neurogenic pulmonary edema in rats was more pronounced in pentobarbital-anesthetized (60 mg/kg) rats compared with xylazine-ketamine anesthesia22 and that it developed in rats anesthetized with 1.5% isoflurane, but not in those anesthetized with 3% isoflurane,20 illustrating that anesthetic drugs used in experimental neurogenic pulmonary edema may be an important factor in the genesis of edema. In the present study, all animals were anaesthetized by ether. However, the influence of anesthetic was not evaluated or compared.
NPE has been also associated with pulmonary vascular permeability.17 Concerning the mechanism of involvement of NO in the maintenance of liquid balance in isolated rabbit lung, it was found that inhibition of NO production with L-NAME caused a significant increase in the rate of weight gain, while the addition of lodoxamide or 8 Br-cGMP attenuated this increase.23 These findings suggest a role for endogenous NO in the relaxation of endothelial cells and also in helping to prevent pulmonary edema by stabilizing mast cells.
Thus, NO production mediated by iNOS has been considered to exert a toxic effect on the pulmonary endothelial layer once L-NAME (10 mg/kg), aminoguanidine (15 mg/kg) or dexamethasone (3 mg/kg) all attenuated endotoxin-induced pulmonary edema.11 As iNOS has been implicated in the pathophysiology of congestive heart failure, it was hypothesized that iNOS deficiency would reduce the severity of congestive heart failure in mice. However, it was found that deficiency of iNOS did not significantly affect severe congestive heart failure in mice after myocardial infarction.24 According to the proposed involvement of NO produced by iNOS mediation on pulmonary edema, it was suggested that morphine had a prophylactic effect on α-naphthylthiourea-induced lung damage through inhibition of iNOS expression.18 The results observed in the present study demonstrate that the inhibition of NO production through iNOS promoted an attenuation of vagotomy-induced pulmonary edema. Thus, it is likely that, in this experimental model, NO produced by iNOS is involved in the development of pulmonary edema. Our group observed a similar result where pulmonary edema induced by endotoxemia was attenuated by aminoguanidine and worsened by L-NAME in control and exercise-trained animals.25
L-NAME increases weight gain in isolated perfused rabbit lung. The authors suggested that NO had a protective role in the maintenance of normal pulmonary vascular permeability.23 Moreover, it was observed that up-regulation of eNOS is a protective mechanism against inflammatory pulmonary damage following ischemia reperfusion.26 Also, the role of NO in the development of NPE in spinal cord-injured rats was considered protective considering that acute treatment with L-NAME (30 mg/kg) just before the injury enhanced NPE severity.20 However, no data in the literature has studied the involvement of both pathways of NO production in neurogenic pulmonary edema.
L-NAME was considered an inhibitor of cNOS, however, it may also inhibit iNOS at a dose of 10 mg/kg.11,27 The treatment of vagotomized rats with 39 mg/kg of L-NAME promoted a reduction in pulmonary edema, that was statistically different when compared with the group treated with 0.3 mg/kg of L-NAME, and the treatment with aminoguanidine (at any dose) reduced the incidence of pulmonary edema in vagotomized rats, as well as in the animals treated with the highest dose of L-NAME. Our hypothesis is that the non-selective blockade of cNOS by administering the highest dose of L-NAME could also inhibit the iNOS pathway. Thus, cNOS appears to participate in the mechanism of pulmonary edema induced by vagotomy, as a protective mechanism. On the other hand, iNOS seems to be directly involved in pulmonary edema induced by vagotomy.