



Non-pituitary tumors have been reported in a subset of patients harboring germline mutations in the aryl hydrocarbon receptor-interacting protein (AIP) gene. However, no detailed investigations of non-pituitary tumors of AIP-mutated patients have been reported so far.
PATIENTSWe examined a MEN1- and p53-negative mother-daughter pair with acromegaly due to somatotropinoma. Subsequently, the mother developed a large virilizing adrenocortical carcinoma and a grade II B-cell non-Hodgkin’s lymphoma.
DESIGNMutational analysis was performed by automated sequencing. Loss-of-heterozygosity (LOH) analysis was carried out by sequencing and microsatellite analysis. AIP expression was assessed through quantitative PCR (qPCR) and immunohistochemistry.
RESULTSThe functional inactivating mutation c.241C>T (R81X), which blocks the AIP protein from interacting with phosphodiesterase 4A (PDE4A), was identified in the heterozygous state in the leukocyte DNA of both patients. Analyzing the tumoral DNA revealed that the AIP wild-type allele was lost in the daughter’s somatotropinoma and the mother’s adrenocortical carcinoma. Both tumors displayed low AIP protein expression levels. Low AIP gene expression was confirmed by qPCR in the adrenocortical carcinoma. No evidence of LOH was observed in the DNA sample from the mother’s B-cell lymphoma, and this tumor displayed normal AIP immunostaining.
CONCLUSIONSOur study presents the first molecular analysis of non-pituitary tumors in AIP-mutated patients. The finding of AIP inactivation in the adrenocortical tumor suggests that further investigation of the potential role of this recently identified tumor suppressor gene in non-pituitary tumors, mainly in those tumors in which the cAMP and the 11q13 locus are implicated, is likely to be worthwhile.
Acromegaly/gigantism is characterized by excess of growth hormone (GH) largely due to GH-secreting pituitary adenomas.1 Although most cases are sporadic, familial forms may occur in association with inherited syndromes such as Multiple Endocrine Neoplasia type 1 (MEN1), Carney complex (CNC), pituitary adenoma predisposition (PAP) and familial isolated pituitary adenoma (FIPA), which includes isolated familial somatotropinoma (IFS).2–4
Germline mutations and somatic inactivation of the aryl hydrocarbon receptor- interacting protein (AIP) gene have been recently identified in patients with PAP.2 The role of AIP in FIPA patients has been confirmed, and more than thirty different AIP inactivating mutations have been identified throughout the gene.5 GH-, GH/PRL- and PRL-secreting pituitary adenomas are the most common clinical features of AIP mutation carriers, although ACTH-secreting and non-functioning pituitary adenomas have also been reported.5,6AIP mutations account for approximately 15% of families with FIPA and 50% of IFS families.3,5 While AIP mutations appear to be very rare in cases with sporadic pituitary disease,7,8,9 they are more frequently found in children and adolescents with GH-secreting tumors, even in the absence of family history.10
Although no systematic clinical surveys of non-pituitary neoplasia have been reported, concomitant non-pituitary tumors, including thyroid, adrenal and MEN1-related tumors were reported in a subset of AIP mutation-positive PAP and FIPA families10,11 and Prof. Albert Beckers, FIPA Meeting, Liège 2009 (unpublished data). The fact that AIP interacts with phosphodiesterases type 4A (PDE4A) and type 2A (PDE2A) implicates this gene in the cyclic AMP (cAMP) signaling cascade,12,13 a cellular pathway known to be disrupted in pituitary, but also in thyroid and adrenal tumorigenesis.14,15 Furthermore, AIP is widely expressed, which may argue in favor of a potential involvement of this gene also in non-pituitary tumors. Thus, one may hypothesize that mutations in AIP may also predispose patients to a broader spectrum of endocrine tumors. However, no genetic investigations of non-pituitary tumors in AIP-mutated patients have been performed to date. In this study, we investigated tumoral samples from a somatotropinoma, an adrenocortical carcinoma and a grade II B-cell non-Hodgkin’s lymphoma from a mother-daughter pair with IFS who harbor a functional AIP germline mutation.
SUBJECTSWe investigated the germline and somatic status of AIP in a previously reported Brazilian family (mother-daughter pair)16 with early-onset acromegaly (at 25 years and at 14 years, respectively) due to invasive pituitary macroadenomas who have been followed in our institution for the last decade. This family has been recently included in the ongoing genetic screening program for Multiple Endocrine Neoplasia at the Endocrine Genetics Unit laboratory at the University of São Paulo School of Medicine.17–24 Since the initial report of a GH-secreting pituitary adenoma, the index case (mother) subsequently developed a virilizing adrenocortical carcinoma and a grade II B-cell non-Hodgkin lymphoma. Because MEN1 and p53 mutations had been previously excluded in this case,16,25 we hypothesized that loss of the AIP gene could be implicated.
CLINICAL FEATURESThe index patient was diagnosed with acromegaly due to a pituitary macroadenoma at 25 years of age (1986). MEN1 and CNC syndromes were ruled out given the absence of the characteristic biochemical and clinical features of these syndromes (such as hyperparathyroidism and enteropancreatic tumors for MEN1 and spotty skin pigmentation, cutaneous myxomas and Cushing’s syndrome for CNC). She underwent transsphenoidal surgery followed by radiotherapy and exhibited persistently normal basal serum levels of GH and insulin-like growth factor-1 (IGF-1) after surgery. At present (48 years old), GH and IGF-1 levels remain within the normal range, and the pituitary MRI was normal, indicating that she had been cured of her pituitary disease.
Thirteen years after pituitary surgery (1999), she presented with virilization and secondary amenorrhea associated with high serum levels of total testosterone (538 ng/dL; normal range, 15–80 ng/dL) and dehydroepiandrosterone sulfate (DHEA-S) (>10,000 ng/mL; normal range, 350–4300 ng/mL). An abdominal CT scan revealed a large heterogeneous mass (9.0 cm x 8.9 cm) in the right adrenal gland compressing the vena cava and the lower portion of the liver. The tumor was surgically excised en bloc without rupturing the organ capsule through a classic lobotomy, and right nephrectomy and extensive resection of local lymph nodes were performed. The histopathological analysis ruled out the invasion of local lymph nodes, liver or vena cava, and the tumor was staged as MacFarlane-II and Weiss-III (high nuclear grade, absence of clear cells and diffuse architecture), indicating adrenocortical carcinoma. There has been no biochemical or imaging evidence of tumor recurrence during 10 years of post-surgical follow-up (1999–2009).
At the age of 42 (2001), the patient developed an apparently indolent grade 2 (WHO classification) slow-growing follicular B-cell non-Hodgkin lymphoma. The immunophenotype was ascertained by immunohistochemical studies of lymph node biopsy samples. The samples were positive for anti-BCL-2, CD10, CD20 and CD68 antibodies and negative for CD3 (in the follicles).
The patient also presented two solid thyroid nodules of 7 mm and 11 mm in size, without adenomegaly, that had been identified in previous cervical ultrasonograms (1999). Pelvic imaging studies (2001) showed ovaries of 5.1 cm and 7.8 cm, respectively, two ovarian cysts (4.7 cm and 5.5 cm) and periaortic, cava-aortic and iliac lymph nodes that were enlarged by up to 2 cm. She underwent a hysterectomy due to adenomyosis and an oophorectomy (right ovary) due to enlarged follicle cysts.
Family historyThe index patient has two daughters (dizygotic twins) and one son (Figure 1A). One of the female twins was evaluated at 14 years of age. She was 167 cm tall and presented with mild acromegaloid features but no signs of gigantism. The GH and IGF-1 serum levels were high, whereas prolactin was normal. An MRI revealed a large pituitary adenoma with suprasellar extension, as well as an invasion of the cavernous and sphenoid sinuses. The tumor was resistant to somatostatin analog therapy. The patient subsequently underwent two transcranial surgeries, one of which involved a transcavernous approach. Histopathological immunohistochemistry analyses indicated that the pituitary adenoma was positive for GH. No clinical and biochemical features of the MEN1 and CNC syndromes were observed in this patient. She has been followed up periodically since 2000, and any adrenal involvement has been ruled out by clinical assessment, determination of adrenal steroid levels and imaging studies (abdominal CT scans). The other female twin and the son were clinically and biochemically normal.
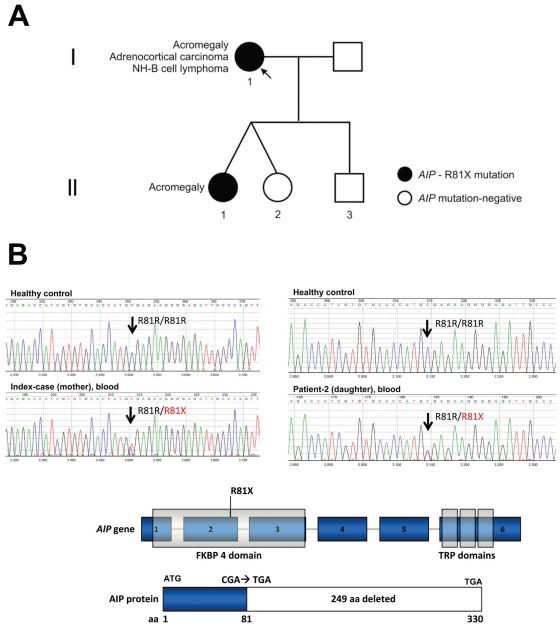
28 was identified in genomic DNA samples from both patients (mother and daughter) with acromegaly but not in the three unaffected family members.")
Genealogy: identification of a functional AIP mutation that may disrupt the cAMP signaling pathway. A - The index patient developed acromegaly, adrenocortical carcinoma and B-cell lymphoma. One of her daughters had acromegaly due to a large and invasive somatotropinoma. B - The known AIP functional mutation c.241C>T (R81X)28 was identified in genomic DNA samples from both patients (mother and daughter) with acromegaly but not in the three unaffected family members.
The index patient reported that one of her sisters also exhibited thyroid nodules and that her mother and another sister had ovarian cysts. These individuals were not available for clinical assessment.
GENETIC (NON-AIP) FEATURESMEN1 geneAs previously reported, the index patient did not harbor a germline MEN1 mutation; however, she and her acromegalic daughter share 13 microsatellite loci at chromosomal region 11q13.16
P53 geneFurthermore, the index patient did not have a germline or somatic R337H TP53 mutation, which is commonly identified in children but less frequently identified in adult patients with adrenocortical neoplasia in southern Brazil.25
TUMORSBoth paraffin blocks and frozen tissue samples of the daughter’s GH-secreting pituitary adenoma and the mother’s adrenocortical tumor and B-cell non-Hodgkin lymphoma were available for study. Samples from the thyroid and ovarian tissues and the mother’s somatotropinoma were not available.
METHODSThe present study was approved by the local ethics committee (Cappesq) of our institution (protocol 0437/08). All patients and control subjects gave written informed consent.
AIP germline mutation analysisGenomic DNA was isolated from the peripheral blood using a standard salting-out protocol, and PCR reactions were performed as previously described.2 Both DNA strands were sequenced from purified PCR products using Big Dye Terminator v3.1 (Applied Biosystems, Foster City, CA) and an automated sequencer (ABI Prism 3130xl DNA Analyser, Applied Biosystems).
Loss-of-heterozygosity analysisTwo different approaches (intragenic and extragenic) were used to verify whether the AIP wild-type allele was lost in the tumors. The mother’s somatotropinoma and thyroid neoplasm were not available for study. We performed PCR amplification, automated sequencing and haplotyping analysis using the D11S1258-11q13 microsatellite marker located close (67,069,747–67,069,958 bp, Ensembl) to the AIP gene 11q13.3 locus (67,007,097–67,015,150 bp, Ensembl).
Quantitative mRNA analysisA commercially available RNA/cDNA pool of normal adrenals (n=61), normal pituitary and normal adrenal tissues obtained from autopsies (Clontech, Palo Alto, CA) were used as controls. Quantitative PCR (qPCR) was performed with an ABI Prism 7700 Sequence Detector using the TaqMan Gene Expression Assays (Hs00610222_m1 for AIP and 43263 for β-actin) following the manufacturer’s instructions (Applied Biosystems). A cycle threshold (CT) value in the linear range of amplification was selected for each sample in triplicate and normalized to the β-actin expression levels. The relative expression levels were analyzed using the 2-δ δCT method,26 where δ δCT is the difference between the selected δCT value of a particular sample and the δCT of a pool of normal adrenals.
AIP immunohistochemistryThe slides were deparaffinized, hydrated and subsequently incubated in a 10 mM citrate buffer (pH 6.0) on a steamer for 40 min at 95 ºC. After several washes, each set of slides was incubated overnight with an anti-ARA9/ AIP monoclonal primary antibody (clone 35-2, Novus Biologicals, Littleton, CO), and then with the Novolink (Vision Biosystems™, Victoria, Au) secondary peroxidase short polymer system. Detailed information on qPCR and immunohistochemistry protocols can be obtained upon request.
RESULTSAIP germline status in the IFS patientsA heterozygous nucleotide change c.241C>T (R81X) in exon 2 of AIP was identified in genomic DNA samples from the two acromegaly patients (mother and daughter) (Figure 1). The R81X is a known functional mutation that codes for a truncated protein lacking the tetratricopeptide repeat (TPR) carboxy terminal domains, which are essential for AIP to bind to the aryl hydrocarbon receptor (AHR) and heat shock 90 (HSP90).12,27 Previous in vitro studies of the R81X mutation have shown that it blocks the interaction of wild type AIP and phosphodiesterase type 4A (PDE4A), potentially disrupting the cAMP cascade.28
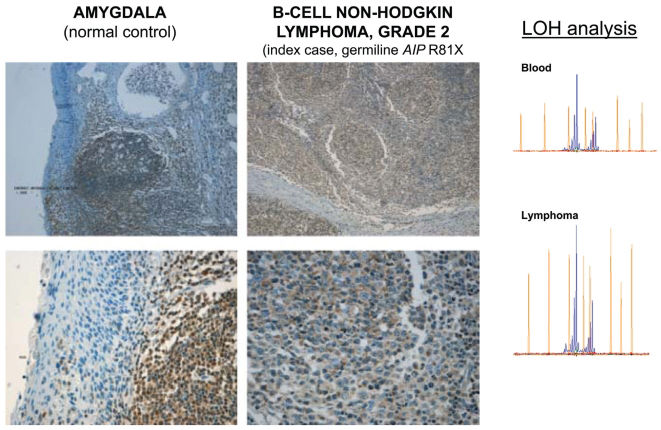
AIP somatic status in the somatotropinoma, adrenocortical tumor and B-cell non-Hodgkin lymphomaSequencing the AIP gene in the tumoral DNA samples revealed a complete loss of the remaining wild-type allele “C” in both the pituitary and adrenocortical tumors (Figures 2B, 3B). Samples from the adrenocortical tumor and B-cell non-Hodgkin lymphoma were analyzed with the AIP-flanking microsatellite marker D11S1258, and LOH was confirmed in the adrenocortical tumor (Figure 3B). However, the lymphoma maintained heterozygosity at the AIP locus (Figure 4).
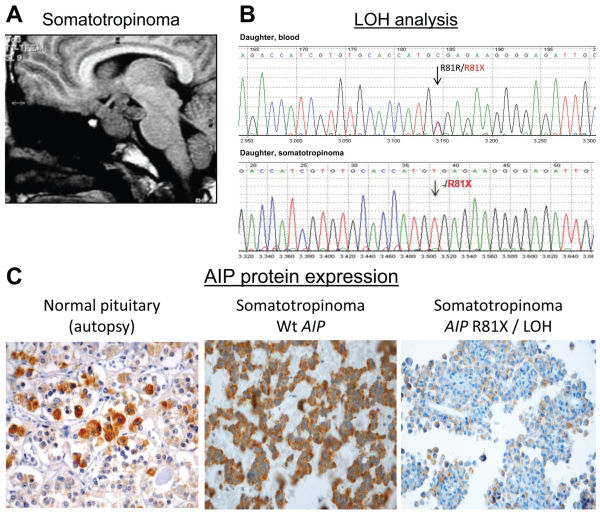
 that was resistant to treatment with a somatostatin analog. She inherited the heterozygous R81X AIP germline mutation (Fig. 1B) from the index case. B - The sequence analysis of AIP in tumoral DNA samples revealed that only the R81X-mutated allele (t) was present in the somatotropinoma, indicating somatic loss and inactivation of wild-type AIP. C - The immunohistochemical analysis showed AIP protein expression in the normal pituitary and a GH-secreting pituitary adenoma of a patient with sporadic acromegaly without AIP mutation. The somatotropinoma of the patient harboring the R81X germline mutation and the somatic loss of the gene presented low AIP protein expression.")
Loss of AIP in the familial somatotropinoma. A - The MRI of the index patient’s daughter revealed a large and invasive pituitary adenoma (so-matotropinoma) that was resistant to treatment with a somatostatin analog. She inherited the heterozygous R81X AIP germline mutation (Fig. 1B) from the index case. B - The sequence analysis of AIP in tumoral DNA samples revealed that only the R81X-mutated allele (t) was present in the somatotropinoma, indicating somatic loss and inactivation of wild-type AIP. C - The immunohistochemical analysis showed AIP protein expression in the normal pituitary and a GH-secreting pituitary adenoma of a patient with sporadic acromegaly without AIP mutation. The somatotropinoma of the patient harboring the R81X germline mutation and the somatic loss of the gene presented low AIP protein expression.
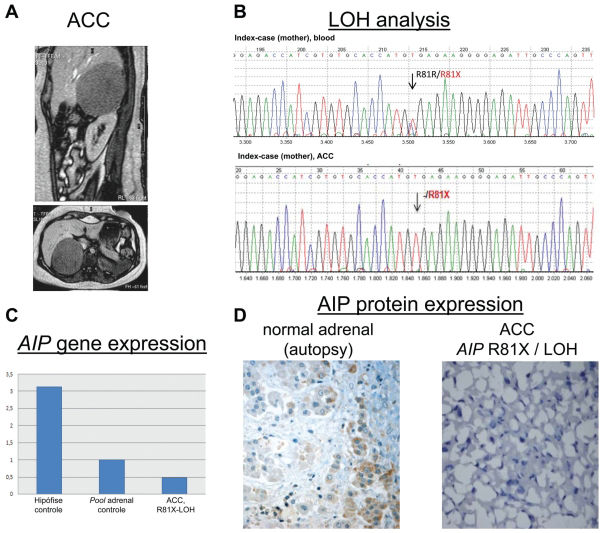
 in the right adrenal gland, which was surgically excised and histopathologically classified as adrenocortical carcinoma. B - Sequence and microsatellite (D11S1258 AIP-flanking marker) analyses of DNA samples from the tumor revealed loss of the wild-type AIP allele. C - The 2-δ δ CT method was used to compare AIP mRNA expression in pooled normal pituitary glands (Ct AIP=25.66, Ct β-actin=24.24), pooled normal adrenal glands (Ct AIP=25.42; Ct β-actin=22.49) and the adrenocortical carcinoma from the index case, in whom the AIP wild-type allele was lost (Ct AIP=31.53, Ct β-actin=27.28). The mean Ct value of the normal adrenal pool was used as a reference (1.0), by comparison, there was decreased AIP expression in the adrenocortical carcinoma (0.48). D - Immunohistochemistry using the AIP antibody showed low/ moderate staining in a normal adrenal gland obtained from autopsy. A complete loss of AIP immunoreactivity was observed in the adrenocortical carcinoma from the index patient, which stained positive for Melan A and 35betaH11, which were used as positive controls.")
Loss of AIP in the adrenocortical tumor. A - An abdominal imaging scan of the index case when she presented with virilization and high serum levels of adrenal steroids. The scan revealed a large heterogeneous mass (9.0 cm x 8.9 cm) in the right adrenal gland, which was surgically excised and histopathologically classified as adrenocortical carcinoma. B - Sequence and microsatellite (D11S1258 AIP-flanking marker) analyses of DNA samples from the tumor revealed loss of the wild-type AIP allele. C - The 2-δ δ CT method was used to compare AIP mRNA expression in pooled normal pituitary glands (Ct AIP=25.66, Ct β-actin=24.24), pooled normal adrenal glands (Ct AIP=25.42; Ct β-actin=22.49) and the adrenocortical carcinoma from the index case, in whom the AIP wild-type allele was lost (Ct AIP=31.53, Ct β-actin=27.28). The mean Ct value of the normal adrenal pool was used as a reference (1.0), by comparison, there was decreased AIP expression in the adrenocortical carcinoma (0.48). D - Immunohistochemistry using the AIP antibody showed low/ moderate staining in a normal adrenal gland obtained from autopsy. A complete loss of AIP immunoreactivity was observed in the adrenocortical carcinoma from the index patient, which stained positive for Melan A and 35betaH11, which were used as positive controls.

The mean Ct values of AIP and β-actin in the pool of normal adrenal samples were 25.42 and 22.49, respectively, while in the adrenocortical carcinoma of the IFS AIP-mutated case, the values were 31.53 and 27.28, respectively. Using the 2-δ δCT method to determine relative expression, we observed decreased AIP expression in the adrenocortical carcinoma in comparison to the pooled normal adrenals (2-δ δCT = 0.48) (Figure 3C). This finding is in accordance with our analysis of the somatic status of AIP, which showed LOH in this non-pituitary (adrenocortical) tumor, as shown in Figure 3B.
AIP immunohistochemistryPituitary tissueDecreased AIP immunostaining was observed in the pituitary adenoma from the daughter harboring the R81X germline mutation, while intense cytoplasmic AIP positivity was observed in the normal pituitary glands obtained from autopsy samples and in a somatotropinoma from a patient with sporadic acromegaly without the AIP germline mutation (Figure 2C).
Adrenal tissueThe mother’s adrenocortical carcinoma showed positive staining for Melan A and 35betaH11 (data not shown), which are two markers of adrenal cortical differentiation used as positive controls, and complete loss of AIP immunoreactivity (Figure 3D). Moderate and strong staining of AIP-positive cells was observed in normal adult (Figure 3D) and infant adrenal tissue, respectively.
Grade II B-cell non-Hodgkin lymphomaNormal AIP protein expression was observed in the index patient’s B-cell lymphoma when compared to the amygdala lymphoid tissue, which was used as a control (Figure 4).
DISCUSSIONRare extra-pituitary tumors have been identified in PAP/ FIPA patients harboring AIP mutations. Mutant AIP cannot interact with cAMP-specific PDE4A and therefore is likely to disrupt the cAMP signaling pathway.12,27,28 This pathway has been previously implicated in both pituitary and non-pituitary tumorigenesis through defects in the PRKAR1A,29GNAS1,30PDE11A31 and PDE8B32 genes. Thus, further investigation of the role of AIP in these concomitant non-pituitary tumors is warranted. Here, we present the first somatic investigations of pituitary and non-pituitary tumors (somatotropinoma, adrenocortical and grade II B-cell non-Hodgkin lymphoma) in a family harboring an AIP germline mutation.
We initially found that both the mother and daughter with acromegaly harbored a functional cAMP pathway-disrupting germline mutation in AIP (c.241C>T; R81X). Through sequence analysis of tumoral samples, we showed that the remaining wild-type allele C was lost in the daughter’s GH-secreting pituitary adenoma and the mother’s adrenocortical carcinoma. LOH was confirmed by analyzing the D11S1258 repeat marker located at the AIP locus (11q13.3). In addition, we observed decreased AIP mRNA and protein expression in the adrenocortical tissue when compared to normal controls (Figure 3C and 3D). In contrast, normal AIP protein expression and no LOH for 11q13.3 locus were verified in the B-cell non-Hodgkin lymphoma from the same patient (Figure 4). According to the Knudson two-hit model, these findings confirm that AIP plays a tumor suppressor role in the pituitary, as previously reported,2,3,4,8 and suggest for the first time that AIP may also be implicated in non-pituitary tumorigenesis. Interestingly, previous genetic studies have strongly suggested the existence of an as-yet-unidentified tumor suppressor gene at 11q13 that is implicated in adrenocortical tumorigenesis.33–35 We are expanding the analysis of AIP to a large cohort of Brazilian sporadic adrenocortical tumors.
Of note, a reported female patient harboring a germline nonsense AIP mutation, L210X, developed a broad panel of neoplasias involving the pituitary, adrenocortical and thyroid glands;10 this previous report suggests that the tumor susceptibility was similar to that observed in our index case. The L201X-mutated patient presented with secondary amenorrhea at the age of 25 and was diagnosed with acromegaly at 27 years of age. Subsequently, she developed a 19-mm non-secretory adrenocortical adenoma and a 4-cm thyroid adenoma. Georgitsi et al. studied 91 cases of “MEN1-related” tumors without MEN1 mutation, and they detected 2 cases (2.2 %) with AIP germline mutations.11 However, no details of the clinical manifestation of the mutated patients were provided, and it is not clear if they were FIPA/IFS patients. Recently, patients with AIP mutations and thyroid disorders, including nodular goiters, follicular adenomas and follicular and papillary thyroid carcinomas, have been described (Prof. Beckers, FIPA Workshop 2009, Liège, unpublished data; Drs. Outi Vierimaa and Pasi Salmela, University of Helsinki, Marianthi Georgitsi’s PhD Thesis). In these cases, it is not known whether the non-pituitary tumors occurring in the FIPA cases harboring an AIP mutation were related to the primary germline event. Further molecular investigations in FIPA/IFS patients with non-pituitary tumors would improve our knowledge of the tumoral susceptibility caused by AIP germline defects.
In conclusion, our study presents the first molecular analysis of non-pituitary tumors in patients harboring the AIP germline mutation. The finding of somatic AIP inactivation in the adrenocortical tumor of a AIP-mutated patient suggests that we should further investigate the role of this recently identified tumor suppressor gene in non-pituitary tumors, especially those tumors in which cAMP and the 11q13 locus are implicated.