




Long-term potentiation and long-term depression are enduring changes in synaptic strength, induced by specific patterns of synaptic activity, that have received much attention as cellular models of information storage in the central nervous system. Work in a number of brain regions, from the spinal cord to the cerebral cortex, and in many animal species, ranging from invertebrates to humans, has demonstrated a reliable capacity for chemical synapses to undergo lasting changes in efficacy in response to a variety of induction protocols. In addition to their physiological relevance, long-term potentiation and depression may have important clinical applications. A growing insight into the molecular mechanisms underlying these processes, and technological advances in non-invasive manipulation of brain activity, now puts us at the threshold of harnessing long-term potentiation and depression and other forms of synaptic, cellular and circuit plasticity to manipulate synaptic strength in the human nervous system. Drugs may be used to erase or treat pathological synaptic states and non-invasive stimulation devices may be used to artificially induce synaptic plasticity to ameliorate conditions arising from disrupted synaptic drive. These approaches hold promise for the treatment of a variety of neurological conditions, including neuropathic pain, epilepsy, depression, amblyopia, tinnitus and stroke.
Long-term potentiation (LTP) is a form of activity-dependent plasticity which results in a persistent enhancement of synaptic transmission. LTP has been a source of great fascination to neuroscientists since its discovery in the early 1970′s1 because it satisfies criteria proposed by Donald Hebb for a synaptic memory mechanism in his influential book ‘The Organization of Behavior’.2 Notably, LTP is long-lasting and input-specific (changes can be induced at one set of synapses on a cell without affecting other synapses). The complementary process of long-term depression (LTD), in which the efficacy of synaptic transmission is reduced, shares these characteristics and has also received much attention as a candidate mnemonic process.3,4 Whether LTP and LTD are the actual synaptic processes underlying learning and memory, as most neuroscientists believe, has not yet been definitively resolved.5,6 However, at the molecular level, it is very clear that LTP/LTD and many forms of memory rely upon similar molecular mechanisms. In addition, it has been demonstrated that LTP- and LTD-like changes in synaptic strength occur as a memory is formed at various sets of synapses in the brain, and that these changes can occlude the artificial induction of LTP and can be occluded by the prior induction of LTP.7-13 The debate on the relevance of LTP and LTD to human memory will in all likelihood continue until we can harness these processes to mimic the formation of a memory without prior experience.14
LTP and LTD have another potentially important role in modern neuroscience, and that is the possibility that they may be exploited to treat disorder and disease in the human central nervous system (CNS). A variety of neurological conditions arise from lost or excessive synaptic drive due to sensory deprivation during childhood, brain damage or disease. Manipulation of synaptic strength using various developing technologies may provide a means of normalizing synaptic strength and thereby ameliorating plasticity-related disorders of the CNS. In this review we will discuss clinical applications of LTP, LTD and related forms of synaptic plasticity and the technologies that may allow the erasure and induction of changes in synaptic strength in the human CNS. As a framework for this discussion we will first provide some background on LTP and LTD.
Features of LTP/LTD: animal studiesLTP was originally observed in vivo in the hippocampus of anaesthetized rabbits at synapses between the medial perforant path and granule cells of the dentate gyrus.1 In this study, LTP was induced using a stimulating electrode to induce a brief high-frequency train of action potentials in the afferent pathway, thereby ensuring coincident pre- and post-synaptic depolarization. Recordings of the synaptic response (the population EPSP) evoked in the population of activated granule cells revealed a lasting enhancement of synaptic strength following tetanic (high frequency) stimulation. Subsequent studies have been almost exclusively conducted on rats and mice. Later it was found that low frequency trains of electrical stimulation (1 Hz) can induce LTD in hippocampal and cortical pathways.15,16 Experiments in intact animals allow for assessment of the longevity of LTP in the hippocampus using chronically implanted recording and stimulating electrodes.17 Under these conditions, and using multiple induction tetani, LTP has been observed to last for a year in rats.18In vitro preparations, however, have provided most of the insights relating to the cellular mechanisms of synaptic plasticity. LTP and LTD have been studied throughout the CNS but, most commonly, at Schaffer collateral-pyramidal cell synapses in the CA1 region of the rodent transverse hippocampal slice. 19 This preparation has proved advantageous in several ways, not least because it allows for patch-clamp recordings to be conducted with relative ease, thereby enabling experimental control over membrane potential. This approach has revealed that repeated pairing of single presynaptic stimuli (causing transmitter release) with post-synaptic depolarization is sufficient to induce LTP, bypassing the requirement for high frequency stimulation.20 Furthermore, the concept of spike timing-dependent plasticity (STDP) has been developed following the important observation in other in vitro preparations that the timing of pre- and post-synaptic action potentials (spikes) determines the polarity of synaptic change. Repeated activation of a presynaptic spike followed by post-synaptic spike, within a brief time window of approximately 50 ms, leads to LTP, while the reverse order leads to LTD.21,22 The transverse slice allows for easy placement of stimulating electrodes in clearly defined afferent fibre populations because the dendritic and cell body subfields can be visualized. Independent stimulation of two afferent pathways has revealed that neighbouring synapses can be independently potentiated or depressed. This property of ’input specificity‚ is an important characteristic of Hebbian LTP and LTD4,23 (see figure 1). The same two-pathway approach led to the discovery of another key characteristic of LTP, associativity. LTP is associative because weakly stimulated synapses, which would not ordinarily undergo potentiation because insufficient postsynaptic depolarization is achieved, do so when the weak stimulation is paired with strong, LTP-inducing, stimulation of other synapses on the same cell.24 As initially implied by Hebb,2 associativity of synaptic storage mechanisms might reflect the associative nature of human memory. These three characteristics of longevity, input-specificity and associativity are important, not just because they fulfill criteria predicted of an efficient memory mechanism, but because they provide clues as to the molecular mechanisms underlying LTP and LTD, mechanisms that could potentially be addressed to rectify synaptic malfunction.
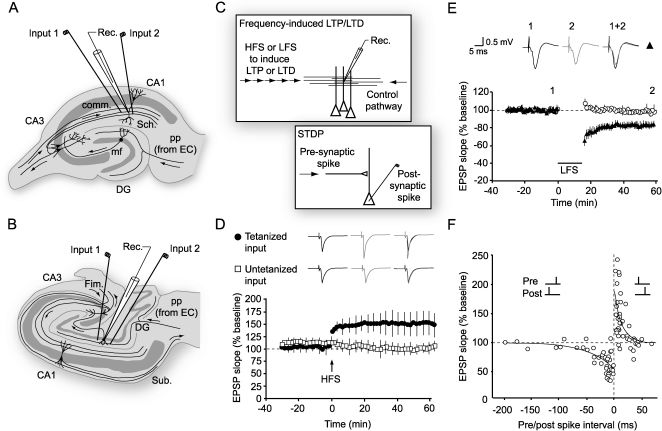
 Schematic of the rodent transverse hippocampal slice, the mostly commonly used preparation for studying LTP, LTD and related phenomena. In the configuration shown, an extracellular recording electrode is placed among apical dendrites of CA1 pyramidal cells, and stimulating electrodes are positioned in the Schaffer (Sch) collaterals to stimulate two separate input pathways. (B) A transverse view of the human hippocampus showing electrode placements for stimulating and recording in the dentate gyrus.67 (C) Diagram showing how LTP and LTD are induced in a pathway (input 1) by high- or low-frequency stimulation (HFS or LFS) respectively, while a second pathway acts as a control (see D,E). A related form of plasticity, known as spike timing-dependent plasticity (STDP), can be induced by repeated pairing of presynaptic and postsynaptic firing. Depending on the relative timing, LTP or LTD is induced (see F). (D) LTP in a two-pathway experiment in the dentate gyrus of human hippocampus. Normalized values of the slope of the population EPSP are plotted for potentiated and control pathways. After a baseline period of test stimulation at 1 stim/min, a brief burst of high-frequency stimulation (HFS, 100 Hz for 1 sec, arrow) is delivered. The potentiation of the response to resumed test stimulation is present immediately after HFS, and persists for at least one hour (black circles) while the control pathway (open squares) remains unaffected, demonstrating the input-specificity of LTP.67 Sample field potentials are shown pre- and post-tetanus. (E) LTD in area CA1 of rat hippocampal slices induced by low frequency stimulation (LFS, ∼1 Hz) for around 15 minutes (bar). This form of plasticity is also input-specific; only the pathway subjected to LFS is depressed (black circles) (adapted from4). (F) The polarity of synaptic plasticity induced by STDP protocols is determined by the relative timing of pre- and post-synaptic spiking. If an action potential is repeatedly evoked before pre-synaptic stimulation then, provided the interval is brief enough, a depression of synaptic strength ensues. If the order is reversed so that pre-synaptic activation precedes the post-synaptic spike, as would happen if the synaptic input contributed to triggering an action potential, then LTP is induced (rat hippocampus, adapted from22).")
LTP and LTD: Induction protocols (A) Schematic of the rodent transverse hippocampal slice, the mostly commonly used preparation for studying LTP, LTD and related phenomena. In the configuration shown, an extracellular recording electrode is placed among apical dendrites of CA1 pyramidal cells, and stimulating electrodes are positioned in the Schaffer (Sch) collaterals to stimulate two separate input pathways. (B) A transverse view of the human hippocampus showing electrode placements for stimulating and recording in the dentate gyrus.67 (C) Diagram showing how LTP and LTD are induced in a pathway (input 1) by high- or low-frequency stimulation (HFS or LFS) respectively, while a second pathway acts as a control (see D,E). A related form of plasticity, known as spike timing-dependent plasticity (STDP), can be induced by repeated pairing of presynaptic and postsynaptic firing. Depending on the relative timing, LTP or LTD is induced (see F). (D) LTP in a two-pathway experiment in the dentate gyrus of human hippocampus. Normalized values of the slope of the population EPSP are plotted for potentiated and control pathways. After a baseline period of test stimulation at 1 stim/min, a brief burst of high-frequency stimulation (HFS, 100 Hz for 1 sec, arrow) is delivered. The potentiation of the response to resumed test stimulation is present immediately after HFS, and persists for at least one hour (black circles) while the control pathway (open squares) remains unaffected, demonstrating the input-specificity of LTP.67 Sample field potentials are shown pre- and post-tetanus. (E) LTD in area CA1 of rat hippocampal slices induced by low frequency stimulation (LFS, ∼1 Hz) for around 15 minutes (bar). This form of plasticity is also input-specific; only the pathway subjected to LFS is depressed (black circles) (adapted from4). (F) The polarity of synaptic plasticity induced by STDP protocols is determined by the relative timing of pre- and post-synaptic spiking. If an action potential is repeatedly evoked before pre-synaptic stimulation then, provided the interval is brief enough, a depression of synaptic strength ensues. If the order is reversed so that pre-synaptic activation precedes the post-synaptic spike, as would happen if the synaptic input contributed to triggering an action potential, then LTP is induced (rat hippocampus, adapted from22).
The transverse slice has the further advantage of allowing the rapid application and removal of pharmacological agents in order to examine their impact on the various phases of LTP and LTD. In this way, the involvement of a wide range of receptors, enzymes and other intracellular signaling molecules in the induction, expression and maintenance of LTP and LTD has been tested. This approach, combined with genetic manipulations in mice to remove or express key proteins, has permitted an extensive characterization of the molecular underpinnings of synaptic plasticity. That LTP requires near simultaneous pre- and post-synaptic depolarization indicates the involvement of a coincidence detection mechanism. It is now known that coincidence detection is accomplished through the NMDA receptor (NMDAR), a voltage-dependent subtype of glutamate receptor that allows permeation of calcium and other cations only when two criteria are satisfied: neurotransmitter is bound following release of glutamate from the presynaptic terminal, and the post-synaptic membrane is sufficiently depolarized to allow the ejection of Mg2+ ions which, at near-resting membrane potentials, block the ion pore of NMDARs.25 Blockade of NMDAR with selective antagonists, such as AP5, CPP or MK801, prevent the induction of LTP but has no effect on its maintenance.26 Genetic manipulation to prevent NMDAR expression in the CA1 pyramidal cell population also prevents the induction of LTP, as well as the expression of several forms of hippocampus-dependent memory.27 Interestingly, this antagonism also blocks the induction of hippocampal LTD,4,28 demonstrating that the two forms of plasticity share a common NMDAR-dependent induction mechanism. Calcium influx through the NMDAR is central to the induction of both LTP and LTD because intracellular application of calcium chelators, such as BAPTA or EGTA, prevents induction of plasticity.28,29 Moreover, uncaging of calcium itself can induce a form of LTP or LTD,30 depending on the concentration of calcium, and both effects are occluded by electrically induced plasticity.31 These are the canonical induction mechanisms for hippocampal LTP/LTD but it is clear that there exist at these synapses and throughout the CNS a wide range of other forms of LTP/LTD that do not rely upon the NMDAR, notably LTD that is induced through activation of the metabotropic glutamate receptor (mGluR).32,33 In addition, the induction protocols that lead to LTP or LTD can vary in different parts of the CNS; for instance, pairing of pre- and post-synaptic depolarization that would lead to LTP in the hippocampus results in LTD at parallel fibre-Purkinje cell synapses in the cerebellar cortex. Purkinje cells are GABAergic and therefore have an inhibitory action on their targets; thus, LTD in this case has a similar net effect to LTP at excitatory principal cells.3
ExpressionA wide range of calcium-detection mechanisms has been implicated in the interface between LTP/LTD induction and expression. Activation of enzymes, such as cyclic AMP (cAMP)-dependent kinase (PKA) and calcium calmodulin-dependent kinase 2 (CaMKII), is essential for induction of the canonical NMDAR-dependent form of LTP in area CA1 of the hippocampus. Both kinases detect elevations in the level of calcium, either directly or indirectly, and are known to phosphorylate proteins that are involved in the expression of LTP, notably AMPA receptors (AMPAR),34 altering their function in ways that enhance synaptic efficacy – for example, by increasing channel conductance.35 Conversely, phosphatases such as calcineurin and PP1, which are also sensitive to calcium but at lower concentrations, can dephosphorylate the same or different protein residues either to reverse LTP, through a process of depotentiation, or to induce de novo LTD, by reducing AMPAR efficacy.36 Thus a complex interplay between the activity of kinases and phosphatases, enzymes which can also either directly or indirectly cross-modulate each others' activity, determines the polarity of synaptic plasticity.37 Persistent activation of these mechanisms initiates a cascade of signaling events that culminate in gene expression and the production of new proteins,38 eventually resulting in much more robust, long-lasting changes in synaptic strength. These protein synthesis-dependent mechanisms underlie the sustained expression of LTP/LTD beyond the first few hours, when the early phase of plasticity (E-LTP and E-LTD) gives way to the late phase (L-LTP and L-LTD). Much of the signaling from the synapse to the nucleus that initiates novel gene transcription is accomplished by a cAMP-dependent signaling cascade involving PKA, mitogen activated protein kinases (MAPK) and the transcription factor cAMP-responsive element binding protein (CREB).39,40 Modulatory neurotransmitters such as dopamine (DA), noradrenaline (NA), serotonin (5-HT) and acetylcholine (ACh), which act on their respective receptors to activate cAMP-dependent signaling in neurons,41-44 also play a role in regulating the longevity of synaptic plasticity. Broadly speaking these neurotransmitters serve as physiological effectors of reward, punishment, arousal and attention, all brain-states that modulate the longevity of memory. Once transcription has occurred the relevant plasticity-related proteins are incorporated, by a process that is not well understood, into just the synapses that are undergoing change and not their neighbours. An interesting theory of synaptic tagging,45 which now has considerable experimental backing,46,47 proposes that a molecular tag is placed at synapses undergoing input-specific plasticity. Newly translated effector proteins are globally expressed and the molecular tag ensures that the relevant effectors are captured only by recently active synapses expressing the tag. The seeming metabolic profligacy of this system may be partially overcome by local translation of existing or newly transcribed mRNAs at ribosomes positioned in the dendrites; local protein synthesis, triggered by a molecular tag, would drastically reduce the number of protein molecules required to potentiate or depress individual synapses.48,49 The molecules that act as tags are not yet determined but there have been some interesting recent proposals, and there is also now a better understanding of what the effector proteins themselves are, as we now discuss.
MaintenanceCaMKII has also been championed as a long-term maintenance mechanism for LTP37 because it can phosphorylate itself and in this way remain autonomously active for a period of time after the dissipation of elevated calcium. This attribute, in principle, would allow it to act as a ‘memorase’.50 Whether or not CaMKII does actually maintain LTP expression for long periods of time has been questioned.51 Inhibitors of this kinase appear to have little effect on already established LTP.52 However, recent data suggests that it may remain active for several minutes after LTP induction. This could enable the kinase to act as a ‘tag’ for recently potentiated synapses and allow for the synapse-specific recruitment of newly synthesized proteins that participate in the maintenance of long-lasting synaptic potentiation.46 Another kinase, PKMζ, has now been recognized to act in a localized fashion to maintain synaptic potentiation for long periods of time. This kinase, an atypical isoform of the calcium-dependent kinase PKC, is remarkable in that it is newly expressed after LTP induction and remains persistently active, in part because it lacks a regulatory domain that would put a brake on its activity in the absence of calcium. Thus, the kinase is capable of maintaining LTP expression at least for its own lifetime and, probably through persistent expression and some as yet not fully understood autoregulation, for much longer periods.53 Indeed, L-LTP can be reversed at least a day after induction through specific inhibition of this kinase in vivo.54 It is now known that PKMζ maintains LTP by increasing the number of AMPARs in the synapse, thereby keeping synaptic transmission potentiated.55 There is abundant evidence that expression of LTP depends on persistent increases in AMPAR number in the synapse,56 although pre-synaptic changes in the probability of neurotransmitter release also play a role, and in some circumstances a dominant role, in supporting at least the early phase of LTP.57-59 The evidence for presynaptic involvement in the expression of LTP in the hippocampus is strong, and has been hard to reconcile with the compelling evidence for changes in glutamate receptor number and/or modification; we are still some way from a unified model of LTP expression.60 LTD also relies on both pre and post-synaptic expression mechanisms61 although here too the maintenance mechanism is not fully understood.
Finally, it is important to mention that structural changes in the size and shape of pre- and post-synaptic specializations may mediate permanent or near-permanent changes in synaptic efficacy. Growth may allow for an increase in the size or number of active zones on both sides of the synapse. Spines can increase in volume after L-LTP induction and decrease after L-LTD induction.62 The degree to which structural re-organisation of synapses occurs in adult animals is not yet clear. An intriguing participant in this later phase of synaptic plasticity is brain-derived neurotrophic factor (BDNF), a substance that is newly synthesised as a result of MAPK and CREB signaling and which may initiate structural change at tagged synapses.63 The role of BDNF in synaptic plasticity is multimodal and it participates in the early phases of both LTP and LTD through co-release with presynaptic glutamate. BDNF, initially in the form pro-BDNF, binds to two postsynaptic receptors: the tyrosine kinase B (TrkB) receptor, whose activation facilitates the induction of LTP,64 and the P75 receptor, whose activation results in an alteration of the subunit composition of the NMDA receptor that promotes the subsequent induction of LTD.65 Thus, BDNF is a major player in synaptic plasticity, although its action is complex (see figure 2).
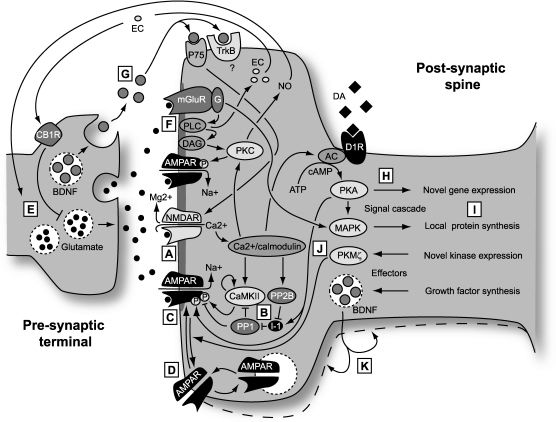
 The induction of canonical forms of both LTP and LTD is triggered by activation of the NMDA class of glutamate receptor. This ionotropic receptor detects the coincidence of presynaptic and strong postsynaptic activity by a mechanism that involves both the binding of transmitter and depolarization-induced repulsion of the Mg2+ ions that block its ionophore at near-resting membrane potentials. In its unblocked state Ca2+ ions are able to permeate the channel, gaining access to Ca2+-dependent processes in the spine and triggering synaptic plasticity. (B) Ca2+ binds to Ca2+/calmodulin which, in turn activates numerous kinases and phosphatases, including CaMKII, PKC and Calcineurin (PP2B) directly and PKA and PP1 indirectly. The balance of kinase and phosphatase activity depends on the concentration and temporal profile of the postsynaptic Ca2+ transient (including Ca2+ released from intracellular stores). The Ca2+ transient determines the polarity of the induced plasticity, with low and prolonged Ca2+ transients inducing LTD and brief, steeper transients inducing LTP. (C) One means by which LTP is expressed is through phosphorylation of the AMPA receptor, an ionotropic glutamate receptor that mediates baseline chemical transmission at excitatory synapses in the CNS. Phosphorylation by CaMKII enhances the conductance of these channels. LTD, by contrast, results, in part, from the dephosphorylation of the AMPA receptor by phosphatases. (D) Trafficking of AMPA receptors plays a major role in the expression of LTP and LTD by increasing or decreasing the number of receptors in the postsynaptic membrane. (E) Presynaptic mechanisms leading to a sustained increase in the probability of transmitter release also contribute to the expression of LTP. The relative contributions of pre- and post-synaptic mechanisms may vary at different times after induction and also across different classes of synapse. Since induction of LTP and LTD is controlled by the post-synaptic NMDA receptor, any presynapic component of expression requires a retrograde messenger that can signal to the pre-synaptic terminal that coincidence has occurred. Two candidates are nitric oxide (NO) and endocannabinoids (EC). (F) A second form of LTD that has been much studied is dependent on group 1 metabotropic glutamate receptors (mGluR). Glutamate binding to this receptor initiates a signal cascade, involving the breakdown of the membrane lipid PIP2 by phospholipase C (PLC) to the important signaling molecules IP3, which releases Ca2+ from Ca2+ stores (not shown) and diacylglycerol (DAG), which leads to the activation of the calcium sensitive kinase PKC. This enzyme then phosphorylates the AMPA receptor but in such a manner that the conductance is reduced. An offshoot is the production of NO. (G) Brain-derived neurotrophic factor (BDNF) plays a complicated role in both LTP and LTD and contributes in different ways to short-term and long-term plasticity. (H) Longer-lasting ‘late’ forms of LTP and LTD, persisting for more than a few hours, require the synthesis of new proteins, either through novel gene transcription or through initiation of local translation of existing transcripts. Novel gene expression requires signaling to the nucleus from newly potentiated or depressed synapses. A major player in this process is the cAMP-dependent signaling cascade initiated by calcium influx and involving adenylyl cyclase (AC) and cAMP-dependent kinase (PKA), which also acts directly on the AMPA receptor in LTP expression. Catecholaminergic modulatory input plays a major role in determining the longevity of LTP and LTD, through interaction with AC which increases levels of cAMP and thereby activates PKA. PKA then sets in action a chain of signals that leads to the expression of new transcripts which, in turn are translated into proteins contributing to the long-term expression of synaptic plasticity. This signaling pathway has been a major recent target of attempts to find nootropic substances. (I) There are parallel signaling pathways, involving mitogen activated protein kinases (MAPK), that also result in the synthesis of new proteins. However, in this case existing transcripts are locally translated into proteins, without further requirement for nuclear signaling. The MAPK pathway is strongly implicated in mGluR-dependent LTD. (J) One newly synthesized protein that acts as a maintenance mechanism for late LTP is PKMζ. This remarkable kinase comprises the active subunit of PKM, an isoform of PKC, that is now known to maintain the presence of AMPA receptors inserted during LTP induction, and thereby maintain LTP. Inhibition of this kinase can erase LTP and memory many days after induction. (K) Finally, BDNF can also play a second role in synaptic plasticity, as a newly synthesized product that alters the structure of the synapse to enforce long-term changes in synaptic strength.")
LTP and LTD: Schematic of molecular mechanisms. (A) The induction of canonical forms of both LTP and LTD is triggered by activation of the NMDA class of glutamate receptor. This ionotropic receptor detects the coincidence of presynaptic and strong postsynaptic activity by a mechanism that involves both the binding of transmitter and depolarization-induced repulsion of the Mg2+ ions that block its ionophore at near-resting membrane potentials. In its unblocked state Ca2+ ions are able to permeate the channel, gaining access to Ca2+-dependent processes in the spine and triggering synaptic plasticity. (B) Ca2+ binds to Ca2+/calmodulin which, in turn activates numerous kinases and phosphatases, including CaMKII, PKC and Calcineurin (PP2B) directly and PKA and PP1 indirectly. The balance of kinase and phosphatase activity depends on the concentration and temporal profile of the postsynaptic Ca2+ transient (including Ca2+ released from intracellular stores). The Ca2+ transient determines the polarity of the induced plasticity, with low and prolonged Ca2+ transients inducing LTD and brief, steeper transients inducing LTP. (C) One means by which LTP is expressed is through phosphorylation of the AMPA receptor, an ionotropic glutamate receptor that mediates baseline chemical transmission at excitatory synapses in the CNS. Phosphorylation by CaMKII enhances the conductance of these channels. LTD, by contrast, results, in part, from the dephosphorylation of the AMPA receptor by phosphatases. (D) Trafficking of AMPA receptors plays a major role in the expression of LTP and LTD by increasing or decreasing the number of receptors in the postsynaptic membrane. (E) Presynaptic mechanisms leading to a sustained increase in the probability of transmitter release also contribute to the expression of LTP. The relative contributions of pre- and post-synaptic mechanisms may vary at different times after induction and also across different classes of synapse. Since induction of LTP and LTD is controlled by the post-synaptic NMDA receptor, any presynapic component of expression requires a retrograde messenger that can signal to the pre-synaptic terminal that coincidence has occurred. Two candidates are nitric oxide (NO) and endocannabinoids (EC). (F) A second form of LTD that has been much studied is dependent on group 1 metabotropic glutamate receptors (mGluR). Glutamate binding to this receptor initiates a signal cascade, involving the breakdown of the membrane lipid PIP2 by phospholipase C (PLC) to the important signaling molecules IP3, which releases Ca2+ from Ca2+ stores (not shown) and diacylglycerol (DAG), which leads to the activation of the calcium sensitive kinase PKC. This enzyme then phosphorylates the AMPA receptor but in such a manner that the conductance is reduced. An offshoot is the production of NO. (G) Brain-derived neurotrophic factor (BDNF) plays a complicated role in both LTP and LTD and contributes in different ways to short-term and long-term plasticity. (H) Longer-lasting ‘late’ forms of LTP and LTD, persisting for more than a few hours, require the synthesis of new proteins, either through novel gene transcription or through initiation of local translation of existing transcripts. Novel gene expression requires signaling to the nucleus from newly potentiated or depressed synapses. A major player in this process is the cAMP-dependent signaling cascade initiated by calcium influx and involving adenylyl cyclase (AC) and cAMP-dependent kinase (PKA), which also acts directly on the AMPA receptor in LTP expression. Catecholaminergic modulatory input plays a major role in determining the longevity of LTP and LTD, through interaction with AC which increases levels of cAMP and thereby activates PKA. PKA then sets in action a chain of signals that leads to the expression of new transcripts which, in turn are translated into proteins contributing to the long-term expression of synaptic plasticity. This signaling pathway has been a major recent target of attempts to find nootropic substances. (I) There are parallel signaling pathways, involving mitogen activated protein kinases (MAPK), that also result in the synthesis of new proteins. However, in this case existing transcripts are locally translated into proteins, without further requirement for nuclear signaling. The MAPK pathway is strongly implicated in mGluR-dependent LTD. (J) One newly synthesized protein that acts as a maintenance mechanism for late LTP is PKMζ. This remarkable kinase comprises the active subunit of PKM, an isoform of PKC, that is now known to maintain the presence of AMPA receptors inserted during LTP induction, and thereby maintain LTP. Inhibition of this kinase can erase LTP and memory many days after induction. (K) Finally, BDNF can also play a second role in synaptic plasticity, as a newly synthesized product that alters the structure of the synapse to enforce long-term changes in synaptic strength.
Many of the mechanisms described above have since been demonstrated to be crucial for LTP in hippocampal tissue excised from humans undergoing temporal lobe surgery to alleviate otherwise intractable epilepsy. In these slice studies, LTP was readily induced in the temporal lobe and at perforant path-granule cell synapses in the dentate gyrus (see figure 1B) using induction tetani comparable to those used in slices from mouse brain.66,67 LTP in human tissue could be blocked by application of the NMDAR antagonist AP5, just as in animal models. Furthermore, artificial elevation of cAMP through application of forskolin, which increases adenylate cyclase activity, results in chemically-induced LTP, a phenomenon that is well documented in rodent hippocampal slices68 and a principal piece of evidence for the involvement of the cAMP-dependent cascade in L-LTP. These studies are important for their verification that the human brain supports an LTP-like phenomenon, but it should be borne in mind that the tissue may have been in a pathological state as it was taken from individuals with an epileptic focus in the temporal lobe. Indeed, slices taken from individuals in whom the focus was present in the hippocampus itself produced far less LTP than those in whom the focus was elsewhere in the temporal lobe. It is possible that LTP was occluded in those slices taken directly from the vicinity of an epileptic focus by previous synaptic potentiation occurring through excessive neural activity. In keeping with this notion, levels of CaMKII expression were found to be significantly higher and levels of the phosphatase PP2B significantly lower in dentate granule cells of individuals with hippocampal epileptic foci.69 Thus, unsurprisingly, it is clear that LTP can be readily induced in the human CNS and that several molecular mechanisms are shared with rodent models. With this in mind we can now consider whether LTP and LTD can be harnessed for therapeutic purposes in humans (see Table 1).
The potential applications of LTP/LTD-like plasticity to the treatment of neurological disorders.
| Disease | Underlying plasticity | Potential Method | References |
|---|---|---|---|
| Depression | Hyper-excitability of prefrontal cortex leading to suppression of activity in target sites. | Low frequency rTMS in prefrontal cortex to induce LTD-like plasticity and ameliorate hyper-excitability of prefrontal cortex. | 163 |
| Parkinson's disease | Reduced nigral drive and resultant loss of function in basal ganglia and motor cortex. | High frequency rTMS in motor cortex to induce LTP-like plasticity and ameliorate lost basal ganglia drive; | 75 |
| Schizophrenia | Hyper-excitability of sensory cortex. | rTMS in auditory cortex to suppress auditory hallucinations with LTD-like plasticity. | 164 |
| Epilepsy | Over-excitability of neural tissue in epileptic focus. | Low frequency rTMS to induce LTD-like plasticity and ameliorate hyper-excitability. | 82,165 |
| Stroke | Lost motor cortical tissue. | DCS in combination with motor task or rTMS to induce BDNF release and promote LTP-like plasticity in surviving cortical circuitry. | 131,135 |
| Chronic pain | LTP-like plasticity in dorsal horn and ACC. | TENS to induce LTD in dorsal horn; rTMS to induce LTD in ACC; Inhibition of PKM to erase LTP in ACC. | 99,103,104 |
| Amblyopia | LTD-like plasticity in primary visual cortex. | Photic tetanus to induce LTP-like plasticity in primary visual cortex; rTMS to primary visual cortex. | 108,121,122 |
| Tinnitus | Over-representation of selected frequencies in auditory cortex due to LTP-like plasticity | Vagal nerve stimulation to cause neuromodulator release coupled with auditory stimulation at lost frequencies to win back incorrectly allocated cortical tissue. | 147 |
Broadly speaking there are two categories of non-invasive approach that can be taken to induce lasting change in neural activity in the human CNS. The first category mimics the high- or low-frequency electrical stimulation used to induce LTP or LTD, respectively. The second removes the requirement for frequency-based stimulation and instead attempts to mimic pairing or STDP-like protocols of induction (see figure 1F and figure 3).
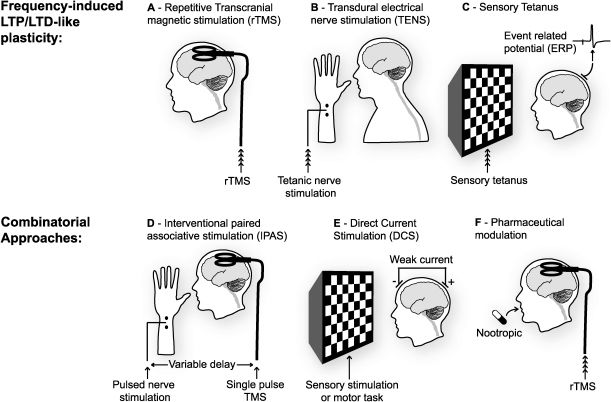
 Repetitive trains of transcranial magnetic stimulation (rTMS), have been employed in several regions of the brain to induce LTP- and LTD-like changes in spontaneous neural activity and responsiveness to stimulation. This approach is limited in the frequencies that can be attained and is restricted to surface structures in the brain, primarily cortical, close enough to the TMS device for effective stimulation. (B) Direct transdural electrical nerve stimulation (TENS) can be used to deliver trains of stimuli to afferent fibres and induce LTP- or LTD-like changes within the spinal cord. (C) Work in primary sensory cortices, notably visual and auditory cortex, has revealed that repetitive sensory stimulation, either flashes or tones, can induce lasting LTP- or LTD-like changes in event-related potentials (ERPs) recorded in these regions. (D) Approaches which combine two of the techniques described above (A-C) can be used to induce changes in synaptic strength that often have the added advantage of reducing the amount of stimulation required and restricting the focus of the effect. Interventional paired associative stimulation (IPAS) mimics STDP protocols by combining rTMS with TENS, or sensory stimulation, to induce lasting changes in neural responsiveness. Just as in STDP, the timing of the two stimuli, in this case one peripheral and one central, determines the polarity of change. (E) As well as TMS, direct current stimulation (DCS) can be used to depolarize neurons in the cortex non-invasively. This technique delivers a very weak current continuously over an extended period. There is some indication that DC stimulation in itself can result in lasting changes in neural responsiveness. However, there is substantial evidence that when be combined with tetanic stimulation, or a sensori-motor task, long-lasting changes in synaptic strength can be induced. (F) Another less temporally defined means of modulating the effects of any direct stimulation of the brain is through the ingestion or injection of drugs. Thus far there has not been a great deal of work on combining nootropics with sensory or direct brain stimulation but this is surely an avenue that is ripe for exploration.")
Non-invasive methods of inducing synaptic plasticity in the human CNS. (A) Repetitive trains of transcranial magnetic stimulation (rTMS), have been employed in several regions of the brain to induce LTP- and LTD-like changes in spontaneous neural activity and responsiveness to stimulation. This approach is limited in the frequencies that can be attained and is restricted to surface structures in the brain, primarily cortical, close enough to the TMS device for effective stimulation. (B) Direct transdural electrical nerve stimulation (TENS) can be used to deliver trains of stimuli to afferent fibres and induce LTP- or LTD-like changes within the spinal cord. (C) Work in primary sensory cortices, notably visual and auditory cortex, has revealed that repetitive sensory stimulation, either flashes or tones, can induce lasting LTP- or LTD-like changes in event-related potentials (ERPs) recorded in these regions. (D) Approaches which combine two of the techniques described above (A-C) can be used to induce changes in synaptic strength that often have the added advantage of reducing the amount of stimulation required and restricting the focus of the effect. Interventional paired associative stimulation (IPAS) mimics STDP protocols by combining rTMS with TENS, or sensory stimulation, to induce lasting changes in neural responsiveness. Just as in STDP, the timing of the two stimuli, in this case one peripheral and one central, determines the polarity of change. (E) As well as TMS, direct current stimulation (DCS) can be used to depolarize neurons in the cortex non-invasively. This technique delivers a very weak current continuously over an extended period. There is some indication that DC stimulation in itself can result in lasting changes in neural responsiveness. However, there is substantial evidence that when be combined with tetanic stimulation, or a sensori-motor task, long-lasting changes in synaptic strength can be induced. (F) Another less temporally defined means of modulating the effects of any direct stimulation of the brain is through the ingestion or injection of drugs. Thus far there has not been a great deal of work on combining nootropics with sensory or direct brain stimulation but this is surely an avenue that is ripe for exploration.
rTMS delivers relatively small electrical currents generated by fluctuating magnetic fields administered over the skull using a figure-of-eight magnetic coil. In most cases it only allows for the excitation of neural circuitry in structures relatively close to the brain surface. For this reason the technique has primarily been used in the neocortex and is not a suitable substitute for the kind of invasive deep brain stimulation (DBS) currently used to treat Parkinson's disease.70 Single TMS pulses evoke event-related potentials (ERPs) that can be recorded using scalp recording electrodes. Importantly, delivery of a high frequency train of rTMS pulses can induce lasting potentiation of ERP amplitude and low frequency rTMS can have the opposite effect. In primary sensory cortex, ERPs can be generated by discrete, transient sensory stimuli within the modality of interest. These ERPs also undergo amplitude potentiation as a result of tetanic stimulation with rTMS.71
rTMS is being assessed as a potential treatment for a number of neurological disorders. Parkinson's disease is a common neurodegenerative disorder that results from the specific loss of dopaminergic cells in the substantia nigra (s.n.) leading to tremor, and impairment of movement initiation and termination. Deep brain stimulation of subthalamic nuclei is a well established treatment in advanced cases of Parkinson's disease, and can be very effective.72 rTMS has been suggested as an alternative, non-invasive treatment. Although the primary locus of dysfunction in Parkinson's is too deep for standard rTMS there are secondary effects that can be addressed with rTMS. When cells in the s.n. degenerate, a conspicuous beta-frequency synchronized activity arises in primary motor cortex (M1) that is believed to contribute to limb rigidity and akinesia.73 In an established animal model of Parkinson's disease, in which primates are treated with MPTP, a substance which selectively kills dopaminergic cells in the s.n., this synchronization of activity in M1 occurs in addition to all the classic motor symptoms of Parkinson's disease. Delivery of high-frequency rTMS (130 Hz) to M1 in these monkeys induces a lasting amelioration of rigidity and akinesia.74 Promisingly, rTMS has also been applied to motor cortex in human Parkinson's sufferers to produce significant but short-lasting improvements in motor performance.75 This example illustrates the important point that, even if the primary area of dysfunction in the CNS is not accessible to non-invasive stimulation, secondary dysfunction in more superficial regions can be targeted in order to ameliorate the resulting behavioral abnormality.76 It is worth noting, though, that a very recently developed technology, ‘deep’ rTMS77 may soon permit non-invasive stimulation of deep targets such as the thalamus, basal ganglia and brainstem nuclei without delivering dangerously large currents to the overlying cortex. This technique, which uses a three-dimensional magnetic coil system that surrounds the cranium in order to generate activity at much greater depths than the traditional figure-of-8 system, may come to replace DBS as a non-invasive treatment for Parkinson's disease. It may also be of benefit in the treatment of obesity, Alzheimer's disease and depression.
A long standing treatment for major depression has been electroconvulsive therapy (ECT). Although still one of few effective treatments for this troubling disorder, it is only used when antidepressants fail because it requires anaesthesia and carries the risk of neural damage. One effect of ECT may be to reduce cortical excitability and, thereby, reverse increases in the excitability of cerebral cortex, notably within prefrontal cortex, during treatment-resistant depression.78 Two explanations have been offered for pathological increases in excitability - a persistent decrease in inhibitory tone, or a persistent potentiation of excitatory synapses as a result of an LTP-like process. ECT results in the expression of several biochemical markers of LTD in animals79 suggesting that ECT causes induction of de novo LTD – or depotentiation of already potentiated synapses - in the cortex. Low frequency rTMS, thought to induce LTD-like plasticity in humans, has been applied to the prefrontal cortex to achieve results similar to ECT in depressive patients.80 rTMS may, therefore, eventually serve as a more controlled and focused alternative to ECT, one which does not require anaesthesia and can be used over multiple sessions without fear of major brain damage.
The fact that low frequency rTMS can be used to reduce cortical excitability, perhaps through LTD-like plasticity in humans71,81 suggests that it may also be a potential treatment for epilepsy. Epilepsy results from hyperexcitability, perhaps through excessive LTP at glutamatergic synapses, or through other mechanisms such as increased intrinsic excitability or reduced inhibition. Thus, LTD-like plasticity, induced with low frequency rTMS, may provide a means to reverse LTP at saturated synapses in order to reduce circuit excitability. Low frequency (0.3 Hz) rTMS was first used as a treatment for epilepsy in an open-case study performed over a decade ago, resulting in a significant reduction in the incidence of seizures for up to a month after five days of treatment.82 Subsequent double-blind randomized trials have produced mixed results,83,84 and it is clear that further work is required to identify more precisely the type of epilepsies that respond to rTMS and to optimize the parameters of stimulation in different cases. Generalized seizures may be more responsive than focal seizures, particularly where the focus is in deep brain structures.
The potential of rTMS to address epileptic foci has brought to the fore the importance of navigation. The efficacy of rTMS can be greatly increased if it is coupled with imaging techniques such as functional MRI (fMRI) to map the affected area, or by systematically recording the effects of stimulation on ERP amplitude or motor output.85 This approach enables the tailoring of treatment to individual cases. For disorders such as Parkinson's disease it is very clear which structure in the brain is the primary site of pathology and, although there are slight individual differences in brain shape, it is relatively straightforward to target the region to be stimulated with DBS. In the case of stroke, a condition in which rTMS application to primary motor cortex holds promise as a treatment, the region of major dysfunction must be tracked in the individual before TMS is applied because each stroke victim has a unique pattern of damage. Neuro-navigated TMS maps motor evoked potentials (MEPs) and the motor threshold for eliciting responses from the muscles of interest to optimise TMS delivery points in individual subjects.86,87 High frequency rTMS can then be used to enhance motor output in affected regions. A systematic study of the efficacy of neuro-navigated TMS as compared to conventional stereotaxically-guided TMS, targeting the representation of the dorsal interosseous muscle of the hand and evaluating the impact of rTMS on reaction time and pinch force, revealed significantly greater efficacy for navigated TMS in both these behavioral outputs as well as in potentiating MEPs and reducing motor threshold.88
In summary, rTMS holds promise in the treatment of a variety of neurological disorders, perhaps through the induction of LTP and LTD-like plasticity. Advances in technology, allowing reliable, safe stimulation of deep structures and careful individual tailoring of stimulation sites and protocols using neuro-navigated TMS, will only expand this potential (Figure 3A).
Transcutaneous electrical nerve stimulation (TENS)Electrical stimulation can also be delivered directly to peripheral nerves through the skin to induce LTP-like and LTD-like changes in central neural responsiveness. Certain clinical conditions result from pathology just a single synapse from the periphery and may be addressed with TENS. Chief amongst these conditions is hyperalgesia. Plasticity at two locations contributes to this chronic pain condition which arises from repeated peripheral application of a noxious stimulus to the periphery. The first occurs at the point of nociception itself (peripheral sensitization); and the second at the first central synapses in the pathway within the dorsal horn of the spinal cord (central sensitization).89 High frequency electrical stimulation can be delivered to nociceptive C fibres in animal preparations90,91 to induce a lasting form of LTP within the dorsal horn. Alternatively, LTP can be induced at these same synapses through natural stimulation of peripheral nociceptors with heat, the noxious chemical formalin or mechanical pinching of the skin.92 Some of this potentiation is homosynaptic and confined to the stimulated nociceptive (C) fibres but there is also a component of heterosynaptic potentiation that enhances transmission at other, surrounding C fibres, and may contribute to secondary hyperalgesia. In addition, sensory (A) fibres that do not mediate nociception may also be potentiated heterosynaptically by C fibre stimulation, perhaps through changes in the intrinsic excitability of common target cells in the dorsal horn. As a result there can be a lasting sensitization of sensory nerves around the site of injury leading to hypersensitivity to non-noxious stimuli (allodynia).89
Central sensitization has several molecular parallels with LTP in the hippocampus.93 LTP in the C fibre pathway is both NMDAR-dependent and calcium-dependent,94,95 and also relies upon other molecular players such as Substance P and the NK-1 receptor.96 Blockade of the NMDA receptor with the non-competitive antagonist ketamine during surgery in humans helps to prevent central sensitization and reduce subsequent hyperalgesia on recovery,97 and other NMDA receptor antagonists have similar effects,98 Given the evidence for the involvement of LTP in hyperalgesia, as well as other forms of peripheral and central plasticity that increase responsiveness to peripheral stimulation, one clinical approach is to attempt to reverse central sensitization in the dorsal horn with low frequency stimulation of C fibres to induce LTD, or to ameliorate peripheral sensitization by reducing dorsal horn responsiveness with LTD of all afferent sensory fibres. TENS delivered to peripheral nerves containing both A and C fibres can be used to induce LTP or LTD in the dorsal horn. Significantly, these forms of plasticity respectively increase or decrease the perception of pain in human subjects,99,100 so that, for example, pinpricks become less painful after the delivery of low frequency TENS. The effect of inducing LTD is, however, complicated by the fact that, while low frequency stimulation reduces pain sensitivity directly at the conditioning site, it has the reverse effect of secondary hyperalgesia at adjacent sites.99 It is also important to note that low frequency stimulation of C fibres, which mimics a characteristic activity pattern after injury, can induce LTP with similar biochemical hallmarks to that induced with high frequency stimulation.96 In fact, the most effective way to induce LTD of C fibre-evoked responses in the dorsal horn is to stimulate at a low intensity that will activate A fibres specifically and not the C fibres themselves.91 Thus, it is clear that while the use of TENS holds promise for addressing hyperalgesia, the condition is complex and is likely to require additional treatments to ameliorate all its various components. Nonetheless, we regard the use of TENS to treat neuropathic pain as an exemplar for the development of non-invasive clinical strategies based on the induction or suppression of LTP or LTD. The fact that peripheral stimulation can be delivered immediately afferent to the targeted synapses, as well as the availability of good animal models of hyperalgesia, emphasises the potential for a non-invasive strategy based on TENS. Rational drug design based on manipulating the potentiated and/or depressed state of synapses at the first stages of transmission in peripheral nociceptive pathways offers another promising approach.
The application of TENS has largely been limited to situations where stimulation of monosynaptic pathways is possible. Potentiation or depression may occur di or tri-synaptically but this is difficult to monitor, or to predict, as the frequency of stimulation will change due to filtering by afferent circuitry. Nevertheless, it is known that hyperalgesia is associated with changes in the brain as well as those documented in the spinal cord. The cortex, notably somatosensory cortex and anterior cingulate cortex (ACC), has been a site of investigation because peripheral injury, in the form of removal of a digit in rats, induces NMDAR-dependent plasticity in these regions of cortex.101,102 There are at least three synapses between nociceptive inputs and cortical synapses undergoing LTP-like plasticity but it may be that chronic pain arises from a concatenation of pathological synaptic plasticity at multiple synapses throughout the CNS. Indeed, the delivery of high frequency TENS, producing hypersensitivity to tactile stimulation, does lead to a potentiation of ERPs in the cortex100 and delivery of low frequency stimulation with rTMS in the cortex can lead to a temporary relief of chronic pain.103 In a fascinating recent study of hyperalgesia induced by peripheral nerve injury in mice, local inhibition in the ACC of PKMζ a key maintenance molecule for NMDAR-dependent LTP, resulted in reversal both of LTP-like plasticity induced in the ACC by nerve damage and reduction of hypersensitivity to tactile stimuli.104 This latter study opens up another intriguing avenue of treatment for neurological disorders arising from pathological LTP-like plasticity, namely the local and selective erasure of potentiation by suppressing the activity of PKMζ. From a clinical perspective the major problem with this approach is the fact that it would be highly invasive if used in humans and potentially dangerous given the indiscriminate nature of the PKMζ inhibitor in erasing synaptic plasticity and memory. However, there may be other means of selectively erasing LTP such as depotentiation,36,105,106 a phenomenon related to but mechanistically different from LTD. For a limited time after induction, LTP can be reversed, or depotentiated, by low-frequency stimulation, suggesting a potential role for TENS or rTMS in the treatment of pathological plasticity, particularly in cases where the patient can be brought to treatment soon after the causative event.
Photic and auditory tetanizationA series of animal and human studies has demonstrated that plasticity can be induced in primary sensory cortices without electrical stimulation. Here, the electrical stimulus is replaced by rapid sensory stimulation within the modality of interest.107 For example, LTP-like changes, as measured using changes in the amplitude of one component of the visual evoked potential (VEP), can be induced in visual cortex using a photic tetanus (9-13 Hz) that consists of flashing or phase-reversing visual stimuli (for example, checkerboards or sinusoidal gratings) on a computer monitor.108-110 Three important criteria must be met if the location of plasticity underlying this phenomenon is to be securely identified. First, potentiation should be specific to the particular stimulus, so that the potentiation is no longer expressed if the spatial frequency109 or the orientation108 of the stimulus is altered, consistent with plasticity occurring in primary visual cortex, where representations of sensory primitives such as orientation and spatial frequency have not yet been integrated into more complex object representations.111 This latter observation is reminiscent of the well-documented phenomenon of perceptual learning, in which over time subjects become gradually more and more proficient at responding to highly specific visual stimuli,112 and is also consistent with observations made in animals using photic stimulation, in which potentiation of VEPs is both eye-specific and orientation-specific.113 Second, in these animal models it is possible to intervene in the visual pathway and substitute electrical thalamic stimulation for baseline photic stimulation and observe potentiation of the electrically evoked test responses after inducing potentiation using photic stimulation.13,114 The converse is also observed. LTP induced in primary visual cortex of the rat using tetanic electrical stimulation of the thalamus is also reflected in a potentiation of the VEP.13,115 Therefore, the plasticity underlying VEP potentiation does not occur in the retina or in retinothalamic pathways, but in the thalamocortical projection. Finally, it is worth noting that induction of similar plasticity can be prevented through local blockade in visual cortex of AMPA receptor insertion113 or systemic blockade of NMDA receptors,113,114 and expression of LTP can be reversed with local inhibition of PKMζ13 in rodents. Thus, mechanisms that support LTP also support potentiation of VEPs with photic stimulation.
From a clinical perspective, this work may prove useful in treating monocular amblyopia, a prevalent disorder that affects around 1% of individuals worldwide. Amblyopia results from a mismatch in ocular function early in life, leading to preferential allocation of visual cortex to input through the fully functional eye. Many of the ocular dysfunctions that cause this disorder, such as cataracts (lens opacity), anisometropia (mismatched lens refraction) and strabismus (misalignment of the eyes) can be fully repaired. However, if treatment is delayed beyond around 8 years of age in humans, as often occurs for either diagnostic or economic reasons, the cortex will not recover normal responsiveness to input through the previously dysfunctional eye.116 Extensive work in animals has demonstrated that monocular deprivation through eyelid suture, which models the causes of amblyopia in humans, results in LTD of excitatory transmission within visual cortex.117-119 Therefore, an obvious strategy to rescue the effects of amblyopia is to artificially induce LTP at the same synapses. Two treatments that have been shown to have some efficacy in adult amblyopes are perceptual learning120,121 and rTMS,122 although in general functional recovery has been both modest and short-lived. Future studies may make use of high frequency photic stimulation as a means of recovering function through the amblyopic eye (Figure 3C). It is also worth considering combinatorial approaches to inducing LTP; peripheral sensory stimulation, combined with central activation by rTMS, may be more fruitful in inducing lasting plasticity.
Interventional Paired-Associative Stimulation (IPAS)One non-invasive way to achieve an effect akin to spike timing-dependent plasticity (STDP) in the human cortex would be to activate afferent sensory pathways using, for example TENS or visual stimulation, in appropriately timed conjunction with single pulses of TMS to activate post-synaptic neurons at the target site. This approach, known as interventional paired associative stimulation (IPAS), uses the amplitude of the event-related potential (ERP) to determine the point at which peripheral stimulation initiates synaptic release. Central stimulation can then be delivered either before or after this delay in order to induce LTD or LTP-like effects, respectively (Figure 3D). Note that while IPAS, like STDP, depends on the relative timing of pre- and post-synaptic events, the parallel is not exact, since in the case of IPAS polysynaptic pathways are involved. One clear advantage of IPAS over frequency-based methods is that it avoids potential risks associated with the delivery of high-frequency stimulation - there are reported instances of seizure as a result of using high-frequency rTMS.123
IPAS has been applied in both somatosensory cortex and motor cortex, using ERPs in response to stimulation of the median and ulnar nerves as a metric of plasticity in the cortex. Responses of the abductor pollicis brevis (APB) and abductor digiti minimi (ADM) muscles in the hand, to both peripheral stimulation and single cortical TMS pulses, also served as an assay of enhanced or decreased functional output.124 One beautiful aspect of the results from these experiments is the regional specificity of the LTP/LTD-like effects. Appropriate timing of nerve stimulation with somatosensory cortical stimulation can lead to a specific enhancement of ERPs driven in a small partial hand-representation area in cortex without effect on neighbouring representations,125 which contrasts with a broader effect of direct rTMS application. This specificity also holds true for motor output as the muscle response to probe stimulation, either peripherally or cortically, can be potentiated or depressed by appropriate IPAS delays without effect on the neighbouring control muscle.126 The immediate import of this work, from a clinical perspective, may be in addressing focal dystonia, such as writer's cramp, in which chronic spasm or cramping develops in hand muscles, preventing the performance of fine motor skills such as writing. It is unclear whether this disorder arises primarily from cortical or basal ganglia dysfunction but the responses in sufferers of writer's cramp to IPAS were markedly different from normal control subjects in that the specificity of the effect was lost. In contrast to the effects in control subjects, targeting one muscle with IPAS had a comparable potentiating or depressing effect on the other, control muscle, which had not undergone IPAS.127 Thus, a loss of specificity in cortical connectivity may contribute to the dystonia. This example provides an illustration of how non-invasive stimulation techniques in humans may provide information about the aetiology of disorders - almost as important an outcome as the development of a treatment itself.
Direct Current Stimulation (DCS)Another approach uses an older and more basic method to induce plasticity. Direct current stimulation (DCS) applies continuous weak current to a superficial region of the brain using two electrodes fitted on the scalp surface at carefully selected sites. Delivery of this current can have dramatic and lasting effects on ERP amplitude. The polarity of change is determined by the polarity of stimulation.128 From a theoretical perspective, given the careful research that has been conducted in animal models to determine the optimal frequency ranges of trains of discrete electrical pulses for inducing LTP and LTD, and the precise timing of pulses required to induce STDP, the effects of DCS, which is non-pulsatile, are puzzling. It has previously been demonstrated, however, that in rats DCS produces changes in the spontaneous firing rate of cortical neurons.129 Thus, plasticity induced by DCS may be the product of STDP-like interactions between DCS-induced neural activity and discrete stimulation, either in the form of electrical pulses in ex vivo slices,130 or, in humans, rTMS131 or sensory stimulation resulting from a subject's participation in particular tasks.132,133 It is known that DCS causes the release of BDNF and that this substance modulates the induction of NMDAR-dependent LTP through the TrkB receptor.64 Moreover, a combination of DCS and low-frequency stimulation leads to LTP in ex vivo cortical slices.130 Slices taken from mice that do not express BDNF, or in which the TrkB receptor is blocked, fail to show DCS-induced LTP. The fact that low frequencies, which would normally induce LTD, cause LTP when paired with DCS suggests that post-synaptic cells are in a permissive state for LTP as a result of DCS, just as clamping cells in a depolarized state results in low frequency-induced LTP.134
A major application of DCS has been in enhancing motor learning through stimulation of motor cortex, an approach that has great promise in facilitating recovery from stroke by promoting compensatory plasticity in spared motor cortical tissue.133 Interestingly, mice which do not express BDNF have significant deficits in motor learning.130 A common polymorphism (Val66Met) found at a frequency of >30% in the human population is, in animal models, associated with a reduced BDNF concentrations in the synaptic cleft and with impaired motor learning,130 which suggests that a significant minority of the population may suffer from a mild impediment in motor skill acquisition. Remarkably, humans possessing this polymorphism show a reduction in motor cortical plasticity, measured using ERPs, induced by rTMS, IPAS or DCS/rTMS applied to motor cortex.131,135 Thus, there is some mechanistic understanding of the effects of DCS and why it may impact LTP/LTD. DCS is a potentially important tool for clinicians attempting to modulate synaptic plasticity to ameliorate neurological disorders, particularly in the motor system (Figure 3E).
Vagal nerve stimulationVagal nerve stimulation (VNS) is a related technique to TENS, but is more invasive. Direct electrical stimulation requires a minimal surgical procedure in which electrodes are wrapped around the left vagal nerve in the neck.136 Stimulation of the vagus causes, indirectly, widespread release of neuromodulatory substances such as acetylcholine, noradrenaline and dopamine through activation of the nucleus of the solitary tract.137 Release of acetylcholine from neurons originating in the nucleus basalis promotes the induction of plasticity in the cortex.138-141 Several studies have shown that coincident occupation of cholinergic receptors with stimulation of glutamatergic pathways enhances the induction of LTP/LTD.44,142-144 VNS serves as a minimally invasive, and therefore clinically viable, means of stimulating the nucleus basalis and other brainstem structures to induce release of acetylcholine and other neuromodulators. Thus it can be paired with sensory stimulation, task performance or rTMS in a combinatorial approach to maximize recuperative cortical plasticity and enhance its longevity. A recent study used this strategy to address the prevalent neurological condition of tinnitus. The manifestation of this disorder is a distracting, continuously detected tone in the absence of an external auditory stimulus. There are a multitude of causes for tinnitus, including dysfunction of the sensory apparatus itself. However, it is hypothesized to result, in part, from excessive cortical representation of a small range of sound frequencies to which the individual has previously been heavily over-exposed,145 and application of rTMS to this region has had short-lived beneficial effects.146 Induction of tinnitus in rats by repeated exposure to a loud auditory stimulus in a fixed range of frequencies leads to an over-representation of these frequencies in primary auditory cortex and a consequent under-representation of the remainder of the normal frequency range. Thus, LTD-like plasticity of synapses in the area of over-representation and LTP-like plasticity in the areas of under-representation could be used to redress the balance and, potentially, treat tinnitus. This goal has recently been accomplished in rats by pairing VNS with auditory stimuli of the appropriate frequencies to reverse physiological correlates in auditory cortex and restore detection of a full range of auditory stimuli.147 The same approach could in principle be used to treat a range of disorders that arise from cortical misrepresentation through deprivation or over-exposure.
PrimingAnother important feature of experience-dependent plasticity that has been the subject of a great deal of basic research is metaplasticity. This is the phenomenon whereby the past activity of a synapse alters its susceptibility to future plasticity. We will not describe the many forms of metaplasticity that exist (see148 for review) but will briefly mention one form of metaplasticity that presents itself as an ideal candidate for augmenting the effects of non-invasive brain stimulation: priming of LTP. In a typical example of priming, a brief 5 hz tetanus, which in itself does not alter the strength of synapses, greatly enhances the magnitude and duration of LTP induced by a standard 100 Hz tetanus at the same synapses, provided this is delivered within 30 minutes or so of the priming tetanus.149 It is believed that this process results from mGluR-dependent signaling, leading to an increase in intrinsic excitability of cells. Subsequent induction of LTP then becomes more effective because of the increased likelihood of action potentials and, therefore, post-synaptic depolarization for a given synaptic input.150 Priming of this sort also increases the longevity of LTP, by inducing a burst of local protein synthesis, making new proteins available for subsequent incorporation into synapses undergoing LTP.151 Priming can be achieved in the human CNS by brief application of DCS or a train of relatively low frequency rTMS to the region of interest prior to the induction of LTP-like plasticity with rTMS or motor training.152-155 These studies suggest that the exploitation of metaplasticity may prove valuable in maximizing the clinical effects of non-invasive stimulation techniques.
NootropicsThe presumed importance of LTP/LTD in mnemonic processes has prompted the launch of a number of drug discovery programs by the pharmaceutical industry aimed at improving memory function in humans and treating disorders of memory such as Alzheimer's disease and other forms of dementia. A variety of drugs, referred to as nootropics, are under development. These range from AMPA receptor modulators, known as AMPAkines,156 to drugs that increase the level of cAMP and, therefore, the activity levels of PKA and its signaling pathway.157 Substances that enhance the efficacy of neuromodulatory substances such as acetylcholine are currently the major available pharmaceutical treatments for Alzheimer's disease158 (for reviews of nootropics see159,160). Note that nootropics do not themselves induce LTP, but rather modulate natural mnemonic processes. However, as with priming, nootropics could be used to enhance the effects of non-invasive LTP/LTD induction in the treatment of disease. An illustration of this combinatorial approach comes from recent work on erasing fear in sufferers of acrophobia (fear of heights) by invoking the natural process of fear extinction (see161 for review). In these studies, application of the nootropic D-cycloserine, a modulator of NMDAR function, was coupled with the use of virtual reality to mimic the experience of standing in a glass elevator, high above a vertiginous drop.162 The result was a striking loss of generalized fear of heights in individuals receiving both treatments. We can imagine extending this approach by combining the delivery of an effective nootropic with the delivery of rTMS, DCS, TENS, repetitive sensory stimulation or IPAS to enhance any of the developing therapies outlined in this review (Figure 3F).
A note of cautionIn humans we can mimic the experimental protocols of frequency-dependent LTP/LTD with rTMS or TENS, and of spike-timing dependent plasticity with IPAS. However, apart from the few ex vivo experiments that have been conducted in excised human tissue, we do not have a physiological read-out in humans that allows us to be sure that we are truly observing synaptic plasticity. It is important to note that ERPs are not as easily interpretable as, for example, hippocampal field potentials. There are two reasons for this. First, the conditions that give rise to synchronous synaptic currents and hence (field EPSPs) in the hippocampus do not exist in the multilaminar neocortex; second, ERPs are recorded at a distance and not at the site of EPSP generation. Thus, the ERP represents a complex of electrical events; monosynaptic EPSPs, polysynaptic EPSPs and action potentials. As a consequence, a change in the ERP amplitude may reflect synaptic LTP/LTD, altered inhibition or increased intrinsic excitability of the underlying cell population. Other assays, such as motor threshold, are even less illuminating about mechanism. There is a burgeoning body of evidence that suggests commonalities in the molecular mechanisms of synaptic LTP/LTD in experimental preparations and non-invasive induced plasticity in humans. Nevertheless, we should exercise caution in equating non-invasively induced changes in ERP magnitude with synaptic LTP and LTD. For this reason, examples of plasticity in the human brain described in this review have been referred to as LTP-like or LTD-like. It will be of great importance to show that plasticity induced non-invasive in humans exhibits longevity, input specificity and associativity, as these are defining features of homosynaptic Hebbian LTP/LTD. A detailed, if still incomplete, mechanistic understanding of these forms of plasticity has emerged from work on animal models over the past three decades, so that a backdrop of rational drug targets and stimulation parameters is already available. Further work to clarify that rTMS, TENS, IPAS and sensory tetani do indeed induce lasting Hebbian synaptic plasticity, and, if so, to identify the synapses at which it occurs, is required if this vast knowledge base is to be effectively exploited for the non-invasive treatment of plasticity-related abnormalities in the human CNS.
CONCLUSIONSWe have described a number of methods for inducing plasticity in the human nervous system, either non-invasively or with minimally invasive surgery. TENS, rTMS, DCS, IPAS, VNS and sensory tetanic stimulation all hold significant promise as potential treatments for a wide range of neurological disorders. The debate on the physiological relevance of LTP and LTD to human learning will no doubt continue but, in the meantime, analogous processes may be harnessed as tools to recover lost function in the nervous system and to ameliorate the effects of pathological plasticity. These methods could, in the future, be used to rescue deficits by directly addressing the site of pathology or by inducing compensatory plasticity elsewhere. The development of stimulation strategies for the non-invasive induction of plasticity in the human CNS has been guided by basic research into LTP, LTD and STDP and, as we have seen, common molecular mechanisms are beginning to emerge in the human and animal literatures. In the future, a combination of nootropic and amnestic drugs with non-invasive stimulation techniques may emerge as the optimal therapy for plasticity-related disorders of the human CNS.
We would like to thank Dr. Steve Gomperts for reading this manuscript and providing critical comments.