



Sepsis is a major cause of mortality and morbidity in intensive care units. Organ dysfunction is triggered by inflammatory insults and tissue hypoperfusion. The brain plays a pivotal role in sepsis, acting as both a mediator of the immune response and a target for the pathologic process. The measurement of brain dysfunction is difficult because there are no specific biomarkers of neuronal injury, and bedside evaluation of cognitive performance is difficult in an intensive care unit. Although sepsis-associated encephalopathy was described decades ago, it has only recently been subjected to scientific scrutiny and is not yet completely understood. The pathophysiology of sepsis-associated encephalopathy involves direct cellular damage to the brain, mitochondrial and endothelial dysfunction and disturbances in neurotransmission. This review describes the most recent findings in the pathophysiology, diagnosis, and management of sepsis-associated encephalopathy and focuses on its many presentations.
The doctors said that it was puerperal fever andthat it was ninety-nine chances in a hundred it would end in death. The whole day long there was fever, delirium, and unconsciousness. At midnight the patient lay without consciousness, and almost without pulse. The end was expected every minute. Leo Tolstoy's Anna Karenina
Sepsis syndromes, which account for up to 50% of deaths in intensive care units (ICUs), are associated with high morbidity and mortality rates.1 Early recognition and appropriate therapy of sepsis could alter the impending cascade of hypoperfusion, inflammation and organ dysfunction, thereby reducing the mortality rate.1 Most proposed sepsis treatments focus on prompt antibiotic therapy, control of the infectious source, and global hemodynamic goals.2 Organ dysfunction is often diagnosed using laboratory parameters rather than on clinical parameters.3 Brain dysfunction, or sepsis-associated encephalopathy (SAE), has been neglected until recently because there are no precise, well-established clinical or biological markers of damage to the brain during sepsis.4
This narrative review describes the most recent findings in the pathophysiology and diagnosis of SAE, as well as advances in the management of patients with SAE. Because much of the scientific knowledge regarding SAE is relatively new, this comprehensive review covers most of the physiological and clinical aspects of this condition. A directed, non-systematic search of the medical literature using Pubmed, OVID, and Google Scholar was performed to identify research and review articles involving the pathophysiology, diagnosis, and treatment of SAE. The most relevant studies were selected in the following order:
- a.
clinical trials.
- b.
animal models of clinical disease.
- c.
laboratory studies reporting relevant findings on SAE pathophysiology.
Although delirium is a major component of the clinical presentation of SAE, the two conditions are not synonymous. SAE is defined as any brain dysfunction related to septic illness. Delirium is a fluctuating disorder of consciousness that is associated with a change in cognition or perceptual disturbance; it can be caused by any general medical condition.5 SAE is one of many causes of delirium, but delirium is not the only clinical presentation of SAE.
SAE is not a newly recognized disorder. In 1903, Dr. Kingsmill Jones defined febrile delirium as a prejudice of the brain's higher functions triggered by a febrile infectious disease that could occur during or after fever.6 He asserted that post-febrile insanity was a rare condition that was not “merely due to the wasting and exhaustion caused by fever.” According to Dr. Jones' theory, the condition could not be cured with rest and good nutrition because “the specific poison of the fever must be a factor in causing post-febrile insanity.” Dr. Jones was the first author to hypothesize that the same factor that caused fever could be related to brain dysfunction.
Physicians are now aware that SAE is a common condition4,7 that can significantly impact patient outcomes. Altered mental status is present in up to 23% of patients with sepsis.7 Mental dysfunction may even precede the cardinal findings of sepsis.8 When present, SAE is associated with a poor prognosis.7 In the classic report by Sprung et al.,7 the mortality rate of septic patients with altered mental status was 49% compared with a rate of 26% in septic patients with no neurological symptoms. Eidelman et al.9 reported that the severity of neurological symptoms secondary to encephalopathy in patients in the ICU, as assessed by the Glasgow Coma Scale, was correlated to prognosis, with a mortality rate of up to 63% in patients who presented with Glasgow Coma Scale scores between 3 and 8. In a recent epidemiological multicentric study with 497 patients, including 76 septic patients, the overall prevalence of delirium was 32.3%.10 ICU and hospital mortalities were increased in patients with delirium. No studies to date have evaluated the actual incidence of the various types of SAE. Therefore, the actual incidence of neurological changes due to sepsis and its associated mortality might be underestimated.
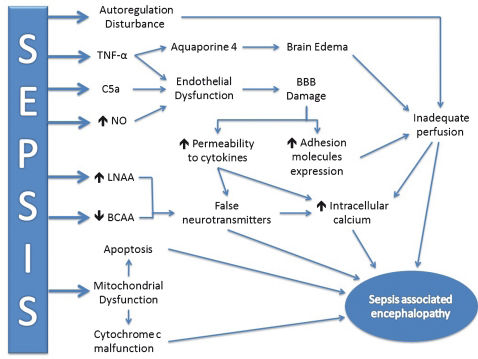
PathophysiologyThe complete pathophysiology of SAE is unknown,11 but several mechanisms have been proposed. SAE appears to involve direct cellular damage to the brain, mitochondrial and endothelial dysfunction, neurotransmission disturbances and derangements of calcium homeostasis in brain tissue.12 Cerebral blood flow may also be affected.13 Because the evaluation of cerebral function during human sepsis is difficult, much of the associated knowledge comes from laboratory studies in animal models of sepsis. Figure 1 summarizes the main mechanisms related to SAE pathogenesis.
 may induce apoptosis and trigger brain injury. The accumulation of false neurotransmitters may increase intracellular calcium content and contribute to encephalopathy. Cerebral perfusion may be altered, rendering brain function more susceptible to injury. TNF-α: Tumor necrosis factor α. NO: nitric oxide. LNAA: Large, neutral amino acids. BCAA: Branched-chain amino acids. SAE: sepsis-associated encephalopathy. BBB: blood-brain barrier.")
Schematic representation of the main pathophysiological mechanisms of SAE. The inflammatory response is represented by TNF-α induction, which increases BBB damage and the liberation of aquaporin 4 to cause brain edema. Mitochondrial dysfunction and the reduction in oxidative phosphorylation efficiency (cytochrome c malfunction) may induce apoptosis and trigger brain injury. The accumulation of false neurotransmitters may increase intracellular calcium content and contribute to encephalopathy. Cerebral perfusion may be altered, rendering brain function more susceptible to injury. TNF-α: Tumor necrosis factor α. NO: nitric oxide. LNAA: Large, neutral amino acids. BCAA: Branched-chain amino acids. SAE: sepsis-associated encephalopathy. BBB: blood-brain barrier.
Neurotransmission: Several neurotransmitters appear to be related to SAE, including the cholinergic pathway14 and the expression of receptors for γ-aminobutyric acid,15 norepinephrine, serotonin, and dopamine.16
Han et al. associated the use of anticholinergic drugs with symptoms of delirium.14 Comim et al.17 evaluated the role of rivastigmine, a cholinesterase inhibitor, in the treatment of memory impairment in rats following cecal ligation and puncture (CLP). Rivastigmine improved performance in an open-field memory test in animals evaluated 10 days after CLP, suggesting that acetylcholine deficiency may be prolonged.
In vivo measurement of neurotransmitters is difficult, requiring lumbar puncture or the use of brain microdialysis.18 Pandharipande et al.18 postulated that the serum measurement of large neutral amino acids (LNAA) could be related to delirium during critical illness because LNAAs are predecessors of many neurotransmitters. Tryptophan (Trp) is necessary for serotonin synthesis; phenylalanine (Phe), together with Tyrosine (Tyr), is required for dopamine and norepinephrine production. In fact, the relationship between plasma Trp and total LNAA (Trp/LNAA ratio) and between plasma Tyr and total LNAA (Tyr/LNAA ratio) were both statistically related to the diagnosis of brain dysfunction during critical illness.18
Reduced levels of branched-chain amino acids (BCAAs- leucine, isoleucine and valine), a group of LNAA, could be related to the increase in brain concentrations of aromatic amino acids (AAA). BCAAs and AAA compete for the same transporter in the blood-brain barrier (BBB).19 If BCAA levels are low, there will be an increase in brain uptake of AAA and, theoretically, an increase in levels of neurotransmitters associated with SAE.
Berg et al.20 further explored the relationship between BCAA and AAA in sepsis. After the infusion of E. coli lipopolysaccharide (LPS) in 12 healthy individuals, the plasma BCAA/AAA ratio declined, mainly because of increases in serum concentrations of Phe and decreases in concentrations of valine and isoleucine. Phe is potentially neurotoxic and can generate false neurotransmitters.21 This study confirms that there is an important interplay between BCAAs and AAA. However, no volunteers developed signs or symptoms of SAE.
Supplementation with BCAAs has been suggested as a therapeutic approach because of their decreased concentrations during sepsis.22 Freund et al.22 demonstrated that survivors of sepsis had higher levels of BCAAs than those who eventually died, suggesting a more adequate energy supply in the surviving patients. In a non-matched interventional group of five patients, the infusion of a parenteral nutritional supplement that was rich in BCAAs reversed SAE. However, randomized clinical trials that include the treatment of SAE with BCAAs are lacking.
Mitochondrial dysfunction: The complex association between mitochondrial dysfunction and target organ damage in sepsis has been extensively reviewed.23–25 The roles of mitochondria reach far beyond energy production and involve diverse metabolic pathways, including cell death.25 Sepsis is associated with mitochondrial dysfunction, which can have remarkable consequences for cells and the health of the host. In brief, early sepsis is associated with a decrease in mitochondrial ATP generation, which is likely mediated by cytokines, reactive oxygen species (ROS), and nitric oxide (NO),23 suggesting an energy deficit that could be related to tissue and organ damage. The mitochondria might trigger cellular apoptosis later in the course of the disease by releasing cytochrome c, for example.25
Although necrosis and apoptosis are present in sepsis, several concerns regarding tissue damage have been raised. In most situations, organ function improves as sepsis is treated, indicating a lack of massive or irreversible anatomic tissue damage. Additionally, one study suggested that apoptosis is a minor feature in the autopsies of septic patients.26 The mitochondria might be associated with an adaptive suspended animation-like state23 in which cells either recover or undergo programmed cell death, depending on the magnitude of the offending event.
Cytochrome c oxidase may play an important role in mitochondrial dysfunction,23 and replacement therapy with cytochrome c has been proposed as a possible treatment for sepsis.27 A recent study by d'Ávila et al.28 evaluated mitochondrial function in brain homogenates of septic mice after CLP. The study showed a reduction in the efficiency of oxidative phosphorylation in the mitochondria and reduced levels of cytochrome c oxidase in the brain following CLP.
The mitochondria are also involved with mechanisms related to neural apoptosis during sepsis, as illustrated by Messaris et al.29 An increase in Bax levels was found in neuronal cells in early stages of sepsis after CLP in rats, primarily in areas with more permissive BBBs, including the hippocampus, choroid plexus, and Purkinje cells. The bax gene is part of the bcl-2 gene family and is associated with regulation of programmed cell death on various biological scenarios.29 Increased levels of Bax likely induce apoptosis by releasing cytochrome c from the mitochondria into the cell cytoplasm, although cytochrome c release was associated with improved prognosis in this study. Therefore, mitochondrial dysfunction may be related to both the induction of neural cell apoptosis and an insufficient energy supply.
Brain cell death: Until recently, it was uncertain whether SAE is associated with irreversible brain lesions.30 Critical illness independently is associated with long-term cognitive impairment (LTCI),31 and a link between severe sepsis and LTCI was recently established in a large cohort of patients.32 Brain involvement in critical illnesses may be a trigger for LTCI or a sign of poor prior brain reserves that is not detectable by screening tests. Additionally, LTCI might be a consequence of brain cell death.33
Brain cell death occurs in SAE, as shown by elevated S100β and neuron-specific enolase (NSE).34 S100β is a calcium-binding protein that is produced mainly by the central nervous system and is released during neuronal or glial injury. Because the normal BBB is impermeable to S100β, elevated serum levels of S100β are a marker of neuronal and BBB damage.
Controversy exists regarding the specificity of S100β and NSE35 in predicting brain injury. Increases in S100β levels in critically ill patients without apparent brain damage have been reported.36 The increases in S100β were not correlated with the severity of clinical encephalopathy in cases of sepsis.37 Therefore, elevations in S100β and NSE levels may be related to critical illness per se or may be markers of minor brain damage caused by inflammation and early organ dysfunction that cannot be otherwise diagnosed.
Calcium homeostasis: An increase in intracellular calcium levels is associated with sepsis-induced brain injury.12 Zhan et al. demonstrated that hippocampal intracellular calcium levels were increased after CLP in rats.12 This increase could be related to ischemia, the presence of excitatory amino acids or a direct effect of cytokines. Altered calcium levels may alter neurotransmitter levels and impair learning, memory and cognitive function in septic patients.
Endothelial and BBB dysfunction: Endothelial dysfunction plays an important role in SAE pathogenesis. Under normal conditions, the BBB protects the brain from a number of insults and creates a tightly regulated microenvironment for neural cells. Important functions of the BBB include the regulation of capillary blood flow and the protection of the brain from circulating noxious substances. The integrity of the BBB is maintained by interactions between astrocytic foot processes, pericytes, and endothelial cells.13 During sepsis, breakdown of the BBB facilitates the passage of neurotoxic factors. Disruption of tight junctions in endotoxemia and detachment of brain pericytes in the hippocampus have been reported.38 Both mechanisms could account for brain edema and BBB dysfunction in patients with SAE. In vitro exposure of mouse brain endothelial cells to the plasma of septic animals induces the production of ROS and NO, which may be related to SAE.39
Inflammation is related to increased BBB permeability to cytokines40 and the expression of adhesion molecules. The activation of leukocytes during sepsis causes these cells to adhere to blood vessels and pass to the tissues in a process mediated by adhesion molecules. There is increased expression of intercellular adhesion molecule 1 (ICAM-1) during septic encephalopathy.41 Monocytes that adhere to the endothelium and are transferred to the surrounding tissues can proliferate and expand the population of perivascular macrophages, contributing to cerebral inflammation.13
In addition to experimental models, BBB dysfunction has been demonstrated in septic patients by brain magnetic resonance imaging (MRI).42 BBB breakdown is more frequent around the Virchow-Robin spaces or is diffuse throughout the white matter and posterior lobes. These findings are consistent with posterior reversible encephalopathy syndrome, which is discussed later.4
Inflammatory mediators and complement system: Of the many inflammatory mediators that are involved in SAE, TNF-α appears to be one of the most significant.13,40,43 Alexander et al.43 evaluated the role of brain TNF receptor-1 (TNFR1) in LPS-treated mice. TNF-α induces neutrophil infiltration of the brain tissue, neuronal cell apoptosis, and brain edema (likely by inducing the expression of aquaporin 4). Mice that are knockouts for the TNFR1 gene showed reduced apoptosis and brain edema.
Jacob et al.44 highlighted the effects of the complement cascade during SAE in LPS-treated mice. Genetically modified mice that overexpressed CR1-related y (Crry-tg), a potent complement inhibitor, had an attenuated inflammatory response, with less apoptosis and cerebral edema, after LPS infusion. In addition, inhibition of complement factor C5a in a CLP model was associated with improved BBB function.45
Cerebral blood flow: Brain ischemia has been described in the autopsy findings of patients following death from septic shock, suggesting that there may be alterations in cerebral blood flow during SAE.46–48 Hypotension and low cardiac output could be related to these findings. Because most available data have been obtained from similar autopsy studies, it is uncertain whether cerebral blood flow reduction is universally present in SAE or is only found in extremely ill patients who eventually die. A myocardial depression in septic patients could be linked to insufficient blood flow to the organs, including the brain.47 Earlier reports also suggest that blood flow may be reduced in the brains of septic patients.49
Pfister et al. evaluated cerebral blood flow using transcranial Doppler in a small series of patients.50 There was no association between SAE and altered brain perfusion that was reported in the middle cerebral artery. However, the autoregulation of blood flow was disturbed in patients with SAE, suggesting that the septic brain is more susceptible to variations in mean arterial pressure.
Other factors that may influence cerebral perfusion in sepsis have recently been reviewed.47 Burkhart et al.47 proposed that cerebral perfusion in sepsis is related to both macro- and micro-hemodynamic features. Concerning microcirculation, flow was dependent on the function of the BBB and the local production of NO and ROS. Taccone et al. explored microcirculation in the septic brain in a sheep model.51 Using video microscopy, the authors showed a profound impairment of cerebral microcirculation in sepsis, which was characterized by a reduction in functional capillary density and the proportion of perfused capillaries. These changes were unrelated to the systemic hemodynamic status.
Lessons from NeuropathologyNeuropathology could supply clues to our understanding of SAE.42,46,48 In a series of 23 patients who died from septic shock, all patients had some degree of brain ischemia.46 Other reported findings included multifocal necrotizing leukoencephalopathy (9%), micro-abscesses (9%), hemorrhages (26%), and hypercoagulability (9%). Another series from the same group evaluated brain imaging resonance in SAE and performed autopsies in selected cases42 with varying results. The findings suggested a predominance of white matter lesions, with swelling and dilation of the perivascular spaces and occasional Periodic acid-Schiff stain (PAS) positive fibrin exudates. There were no signs of direct brain involvement, such as micro-abscesses.
Janz et al.48 evaluated the autopsy findings of seven patients with delirium who eventually died. Hypoxic ischemic injury, which was noted in six patients, was the most common finding. Hippocampal injury was present in five patients. Patients with hippocampal lesions spent an average of 5.8 days in delirium and received an average of 2.2 different analgesics and/or sedatives to control their symptoms. As a result, the authors hypothesized that there is a positive correlation between hippocampal injury and symptoms of delirium.
These findings suggest that white matter and the hippocampus are involved in SAE pathogenesis. This conclusion corroborates the concept that BBB damage is one of the key mechanisms involved in SAE. Hippocampal involvement may be related to the clinical picture of delirium.
Clinical Findings and DiagnosisIt must be clearly stated that SAE is a diagnosis based on exclusion.8 Diagnosing SAE is challenging, especially in chronic critical illnesses. Several medical conditions must be excluded before a diagnosis of SAE can be made, including drug use, central nervous system disorders, and electrolyte disturbances. Most patients in the ICU have multiple risk factors for brain dysfunction. Consequently, the brain dysfunction associated with critical illness is often multifactorial.8,52
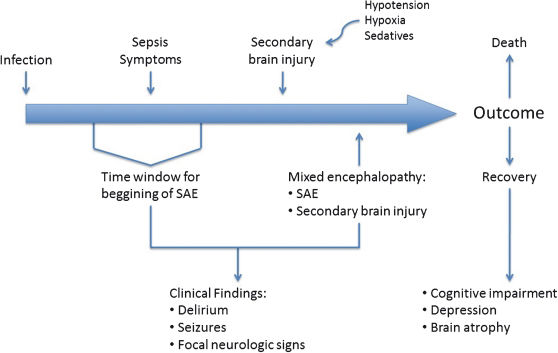
SAE could present with symptoms that range from a few clinical findings to a coma and could entirely bypass the stage of delirium (Figure 2).8,52–54 Therefore, the diagnosis of SAE should not be made using the DSM IV5 criteria for delirium or the CAM-ICU questionnaire.55 To date, no tool is capable of diagnosing the full spectrum of SAE.
. Although SAE may precede other symptoms of sepsis, a diagnosis is usually missing until mixed encephalopathy becomes overt. Outcomes are variable and may be related to the degree of encephalopathy and the occurrence of prior neurological disorders.")
Timeline of sepsis-associated encephalopathy (SAE). Although SAE may precede other symptoms of sepsis, a diagnosis is usually missing until mixed encephalopathy becomes overt. Outcomes are variable and may be related to the degree of encephalopathy and the occurrence of prior neurological disorders.
Altered consciousness is often a cardinal finding of SAE.54 Focal motor deficits and seizures have been reported56,57 but are not common and should prompt a detailed investigation with brain imaging and possibly lumbar puncture. Ordinary causes of coma should also be evaluated.
Electroencephalography (EEG) has been proposed as an important tool in the evaluation of SAE.58,59 Young et al.58 studied 69 septic patients, 49 of whom had some degree of encephalopathy that was categorized as either mild or severe. EEG patterns were classified into five categories: normal, theta, delta, triphasic waves (TW), or suppression. The TW and suppression categories were present only in patients with severe disease. Theta waves were more common in the mild forms of encephalopathy. The EEG pattern was associated with mortality, but the limited number of patients impeded further analysis. However, none of the EEG findings were pathognomonic. EEG may be useful in aiding the diagnosis of SAE because it can exclude non-convulsive status epilepticus.
Brain imaging findings may also be remarkable in patients with SAE.42,60,61 Sharshar et al. performed brain MRI evaluations in nine critically ill septic patients.42 Five of these patients had white matter lesions (leukoencephalopathy) that were characterized by hyperintensity on fluid-attenuated inversion recovery (FLAIR) images and predominated around enlarged perivascular spaces, which are likely related to damage to the BBB. The severity of these lesions was correlated to neurologic prognosis, as evaluated by the Glasgow Outcome Scale.
Posterior reversible encephalopathy (PRE) may also be linked to sepsis and infection.62 In a study of 106 patients with PRE, 23.6% had infection or sepsis/septic shock. The clinical symptoms included altered mental status, seizures and visual disturbances. PRE was not an early finding, as it occurred up to 30 days after infection.
Brain imaging may aid in the diagnosis of SAE and may exclude other causes of neurologic deterioration. Larger case series are necessary to confirm the association between white matter lesions and outcome.
Treating SAETreating and diagnosing SAE are equally challenging tasks. Most treatment schemes are geared toward treating the symptoms of delirium. None of the therapies that aim to treat the pathophysiological mechanisms of SAE have been proven effective in humans.
The use of anti-C5a antibody reduces BBB damage in a CLP model45 in rats, and C5a or C5aR blockade apparently improves mortality in experimental studies.63 The use of drotrecogin alpha has been suggested to reduce neural damage in human sepsis, as evaluated by serum measurements of S100β.64 However, it is unclear whether S100β is an actual marker of brain injury. Because the study reported no clinical endpoints, it is not advisable to advocate the use of drotrecogin in the treatment of SAE.
Sedatives are often used in the intensive care unit, and the use of benzodiazepines is related to the increased incidence of brain dysfunction, as assessed by symptoms of delirium.65 Dexmedetomidine, an alpha-2 agonist, has been associated with less delirium than continuous lorazepam and midazolam in critically ill patients (the MENDS trial).65 Pandharipande et al. performed an a priori analysis of septic patients,66 including 63 patients that were administered either lorazepam (32 patients) or dexmedetomidine (31 patients). Of the two groups, patients who received dexmedetomidine had more delirium/coma-free days (3.2 days; 95% IC 1.1-4.9) and a lower 28-day mortality rate (hazard ratio 0.3; IC 0.1-0.9). This study had several limitations, including possible randomization issues due to the nature of the subgroup analysis and a small sample size, which made it vulnerable to type II error. The results of the study could be related to the beneficial effects of dexmedetomidine or the hazardous effects of benzodiazepines. The possible favorable effects of dexmedetomidine include the inhibition of apoptosis and reductions in the sepsis-associated inflammatory response.66 In the brain, dexmedetomidine has been shown to decrease apoptosis by binding to α2-receptors,67 which may explain the reduction in symptoms of delirium in septic individuals. Until additional evidence is available, dexmedetomidine is preferred over benzodiazepines when sedation is necessary for patients with SAE.
CONCLUSIONSAE is a multifaceted syndrome with serious effects on patient morbidity and mortality. Its pathophysiology is complex and involves hemodynamic and inflammatory features that are not completely understood. Therapy choices are limited and are mainly based on symptom control. Although therapy with antipsychotics is the mainstay of treatment for symptomatic patients for extended periods of time, there are limited data regarding the efficacy and safety of these drugs when administered to septic patients.
Much attention has been paid to symptoms of delirium. The other forms of SAE, including PRE and leukoencephalopathy, have not been thoroughly studied. Furthermore, little is known about the natural history of the disease and the actual impact on quality of life after a critical illness. SAE should not be regarded as an acute reversible state. The brain may be affected in sepsis, similar to any other organ, but with many unique effects. Therefore, efforts should be made to understand the multiple clinical presentations and pathophysiology of SAE to target therapies toward the mechanistic pathways of the disorder rather than to merely control the symptoms of SAE.
No potential conflict of interest was reported.