



Patients with schizophrenia have a two- to three-fold increased risk of premature death as compared to patients without this disease. It has been established that patients with schizophrenia are at a high risk of developing cardiovascular disease. Moreover, an important issue that has not yet been explored is a possible existence of a “cerebral” focus that could trigger sudden cardiac death in patients with schizophrenia. Along these lines, several structural and functional alterations in the thalamic complex are evident in patients with schizophrenia and have been correlated with the symptoms manifested by these patients. With regard to abnormalities on the cellular and molecular level, previous studies have shown that schizophrenic patients have fewer neuronal projections from the thalamus to the prefrontal cortex as well as a reduced number of neurons, a reduced volume of either the entire thalamus or its subnuclei, and abnormal glutamate signaling. According to the glutamate hypothesis of schizophrenia, hypofunctional corticostriatal and striatothalamic projections are directly involved in the pathophysiology of the disease. Animal and post-mortem studies have provided a large amount of evidence that links the sudden unexpected death in epilepsy (SUDEP) that occurs in patients with schizophrenia and epilepsy to thalamic changes. Based on the results of these prior studies, it is clear that further research regarding the relationship between the thalamus and sudden cardiac death is of vital importance.
Patients with schizophrenia have a two- to threefold higher risk of premature death than those without schizophrenia. This increased mortality is accounted for by a combination of factors, such as lifestyle factors, high suicide rates (especially in young male patients soon after diagnosis), premature development of cardiovascular disease, a high prevalence of metabolic syndrome, carbohydrate and lipid metabolic disorders, and, equally important but not often mentioned, sudden unexpected death.1–3 The exact pathophysiological cause of sudden unexpected death in patients with schizophrenia is unknown, but it is likely that cardiac arrhythmias play a role.4,5 It has been established that people with schizophrenia are at a high risk of developing cardiovascular disease and that some atypical antipsychotics may be associated with adverse cardiovascular events (e.g., QT interval prolongation). Together, these risk factors may lead to torsades de pointes or sudden cardiac death.4, 6,7 Moreover, an important issue that has not yet been explored is the possible existence of a “cerebral” focus that could trigger sudden cardiac death in patients with schizophrenia. Studies of the pathophysiology of sudden unexpected death in epilepsy (SUDEP) have revealed that cardiac bradyarrhythmias that are triggered by epileptic activity in the left amygdala, anterior hippocampus, orbitofrontal cortex, and insular cortex can occur and lead to adverse cardiovascular outcomes.8–10 Through their connections with the hypothalamus and pons as well as nuclei in the medulla and spinal cord, these regions can initiate integrated cardiovascular responses related to emotion and motivated behavior that may be inappropriately activated during a seizure.10 The thalamus receives projections from the insular cortex and amygdala and projects to the anterior cingulate cortex, which is the executive region involved in exerting emotional control. This region, in turn, projects to the hypothalamus, periaqueductal grey matter, and the nucleus ambiguus, which control the parasympathetic and sympathetic cardiac ganglia and the atrioventricular node of the heart10. Heart rate, cardiac contractility, and atrioventricular (A-V) conduction may therefore be controlled independently by both these brain structures and the thalamus.11 Thalamic pathology and reduced thalamic volumes have been implicated in the pathophysiology of temporal lobe epilepsy, largely due to the important connection that the thalamus has with the temporal lobe, especially the hippocampus.12 In patients with schizophrenia, thalamic pathology has been described and may be associated with the occurrence of sudden cardiac death. The aim of our paper is to discuss the possible association of sudden cardiac death in schizophrenia with abnormalities in the thalamic complex.
The presence of anatomical abnormalities in the brains of patients with schizophrenia has been well established, especially since the introduction of several imaging modalities that can be used to perform structural and functional brain imaging.13 The structural findings in the brains of patients with schizophrenia include dilatation and enlargement of the ventricles,14 volume reductions in the frontal grey matter,15,16 bilateral reductions in hippocampal volume,17 and a reduction in thalamic volumes. These findings have all been obtained via structural magnetic resonance imaging (MRI) studies.18,19 Because the thalamus is heterogeneous, schizophrenia-related changes would likely occur in specific thalamic subregions (Figure 1). This brain structure is composed of various subnuclei that have received special attention in schizophrenia research due to to their unique locations and neuronal connectivity.13,20–23 The thalamus acts as a central relay station that transfers peripheral sensory input to the cortex, thereby playing a critical role in filtering sensory information, regulating cognitive input to the cortex, and mediating corticocortical connections between areas that have been especially implicated in schizophrenia, such as the frontal and temporal cortices.21,24 Recently, Lang and colleagues25 demonstrated the presence of a bilateral reduction in thalamic volume in a cohort of drug-naive or minimally treated patients experiencing initial episodes of psychosis, suggesting that such abnormalities are present very early in the course of the disease.
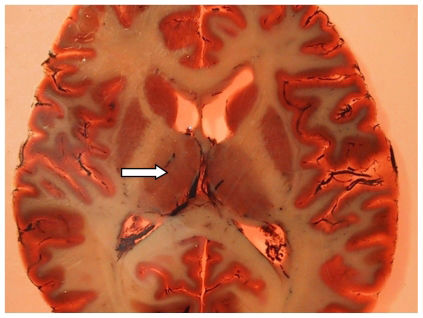
, which is located adjacent to the ventricles. The thalamus is divided into microscopically definable subnuclei (e.g., the mediodorsal nucleus) and has connections to the prefrontal and temporal cortices.")
Photograph of a plastinated brain section from a patient with schizophrenia, showing the situation of the thalamus (arrow), which is located adjacent to the ventricles. The thalamus is divided into microscopically definable subnuclei (e.g., the mediodorsal nucleus) and has connections to the prefrontal and temporal cortices.
The existence of some structural alterations in the brains of schizophrenic patients is supported by data from functional studies that have shown the presence of abnormal thalamic glucose metabolism in these patients.26,27 In patients with schizophrenia, positron emission tomography (PET) studies have shown evidence of dysfunction of the cortico-cerebellar-thalamic-cortical neuronal circuit. This dysfunction contributes to the presence of “cognitive dysmetria”, i.e., impaired cognition, as well as other symptoms of the disease.28,29 Similarly, reduced levels of metabolic activity in the thalamus as measured by PET and single-photon emission computed tomography (SPECT) studies of cerebral perfusion have been linked to cognitive deficits and an increased severity of both positive and negative symptoms in schizophrenic patients30. Functional MRI studies have shown that these patients have sensory processing and attention deficits that involve thalamic hypofunction.31–35
THALAMIC ABNORMALITIES ON THE CELLULAR AND MOLECULAR LEVELOn the cellular level, the density of the parvalbumin-immunoreactive varicosities in the middle layer of the prefrontal cortex has been shown to be lower in schizophrenic patients, suggesting that these patients have fewer neuronal projections from the thalamus to the prefrontal cortex.36 Post-mortem findings of reduced thalamic volume, neuronal number, and overall thalamic size37 as well as reductions in the size of the mediodorsal nucleus, pulvinar nucleus, and ventral posterior thalamic nucleus38–42 support the hypothesis that thalamic function and the connections between various thalamic nuclei and different brain structures in patients with schizophrenia are disturbed. In recent 1H-MRSI (proton magnetic resonance spectroscopy imaging) studies, reduced N-acetyl-aspartate (NAA) levels have been noted to be present in the entire thalamus as well as the anterior and mediodorsal nuclei of the thalamus in patients with schizophrenia.43–45 Reductions in NAA levels indicate a loss of the functional and structural integrity of neurons. However, a correlation between NAA levels and structural parameters has not been demonstrated.42–48 Therefore, alterations of NAA may be related to the dysfunction of neurons with altered glutamatergic neurotransmission.49,50
The glutamate hypothesis of schizophrenia is based on the observation that phencyclidine and ketamine, both of which block the ion channel of the glutamatergic N-methyl-D-aspartate (NMDA) receptor, initiate NMDA receptor hypofunction and precipitate psychosis,51,52 resulting in a final hypoglutamatergic state in the corticostriatal projections.53,54
In the striato - thalamo - cortical loop, this hypoglutamatergic state may lead to a dysfunction of the gamma-amino-butyric acid (GABA)ergic striatothalamic projections with consecutive thalamic disinhibition and glutamatergic overflow to the cortex.24,55 Accordingly, in a 4.0-Tesla MRI spectroscopy study, higher levels of glutamine resulting from glutamate breakdown in glial cells) have been reported to be found in the thalami of unmedicated and chronic schizophrenic patients.56,57 These results correspond to an increased expression of glutamine synthetase and glutaminase. The former converts glutamate to glutamine, and the latter converts glutamate to glutamine. The upregulation of these enzymes suggests an increased rate of glutamate turnover.58 Consistent with these findings, the gene expression of the glial glutamate transporters [excitatory amino acid transporters 1 and 2 (EAAT1 and EAAT2)] that remove glutamate from the synaptic cleft as well as the expression of a vesicular glutamate transporter (VGLUT2) have been shown to be increased in patients with schizophrenia.59,60 On the one hand, this increased expression may occur as a result of transcriptional regulation due to an increased glutamate concentration in the synapse. On the other hand, the increased expression of these transporters may induce a hypoglutamatergic state by decreasing the amount of glutamate that is present in the synaptic cleft. The mRNA expression of the NMDA receptor subunits NR1, NR2B and NR2C, the ionotropic glutamatergic AMPA receptor subunits gluR1 and gluR3, the kainate receptor subunit KA2, and the polyamine and glycine binding sites of the NMDA receptor has been shown to be decreased in the mediodorsal and central medial nuclei of the thalamus in schizophrenic patients.61,62 These changes suggest a diminished glutamatergic activity or abnormal glutamatergic innervation of afferent or efferent cortical or limbic (hippocampal) structures. In contrast, Clinton et al.63 reported the presence of increased protein levels of the NR2B subunit and its associated protein PSD95 (which targets glutamate receptors to the synaptic membrane, modulates receptor activity, and coordinates glutamate receptor-related signal transduction) in the thalami of patents with schizophrenia. These conflicting results are not surprising, since changes in the mRNA levels of a gene transcript may not always reflect alterations in protein levels.
Altered NAA levels may also be related to the decrease in neuropil (dendrites and axons) content that is present in patients with schizophrenia. In concordance with this hypothesis, in a post-mortem study the levels of the synaptic protein rab3a were found to be reduced in the left thalamus of schizophrenic patients.64 Alterations in the neuropil content of specific brain regions may also reflect a reduction in the presence of components of cell membranes, i.e., phospholipids. In a 31P-MRSI (phosphorus magnetic resonance spectroscopy imaging) study, a decrease in the levels of the membrane breakdown product glycerophosphoethanolamine has been observed in the left thalamus of patients with schizophrenia. This change has been viewed as indicative of altered phospholipid metabolism in that region.65 Accordingly, in a post-mortem study, we showed that there was a decrease in the levels of the main membrane phospholipid (phosphatidylcholine) and the major myelin membrane components (sphingomyelin and galactocerebrosides 1 and 2) in schizophrenic patients. In contrast, phosphatidylserine levels were increased in these patients, supporting the concept that there is an increased breakdown of phospholipids in schizophrenic patients. Since these phospholipds are mainly present in the myelin sheath of oligodendrocytes, his finding was thought to be indicative of oligodendrocyte dysfunction and decreased myelination in the thalami of these patients.66
THALAMIC NUCLEAR ABNORMALITIES AND SUDDEN CARDIAC DEATH IN SCHIZOPHRENIC PATIENTS: AN AVENUE TO BE EXPLOREDIn light of the findings mentioned above, the question arises as to whether sudden cardiac death in schizophrenia could be related to functional or anatomical changes in the thalami of these patients.
It has long been recognized that, in the healthy brain, stimulation of (or the presence of lesions in) certain central nervous system structures can lead to morphological and functional cardiovascular alterations. Because it has been documented that disorders of the central nervous system (CNS) can alter cardiovascular function,8 we believe that this issue should be deeply explored in schizophrenia research. Before these studies are undertaken, it is prudent for us to clarify our research background. In a previous study, our research group found that the thalamus plays an interesting and important role in the cerebral circuits of rats with epilepsy67,68. Interestingly, in studies of epilepsy, which is the most prevalent serious neurological condition worldwide, clinical data suggest that these patients are at a two- to threefold higher risk of dying prematurely than those without epilepsy. The most common epilepsy-related cause of death is SUDEP.69
Following this reasoning, we proposed a new line of thinking: because several morphological and/or functional changes of the thalamus have been reported to occur in patients with epilepsy, it is possible that this brain region might also be involved in the occurrence of cardiovascular abnormalities and hence SUDEP.
To answer this question, we initially based our hypothesis on the elegant study performed by Boyko and colleagues.70 The authors showed that bilateral injections of kainic acid into the thalamus of adult rats led to myocardial necrosis. This effect was particularly marked in the lateral posterior thalamic nuclei, suggesting that this specific thalamic nucleus has a direct relationship with the cardiovascular system. Based on these results, we developed an experimental study to reinforce this concept. First, in 2005, we evaluated the heart rate of rats with epilepsy, both in vivo (via electrocardiography) and in an isolated ex vivo preparation (via Langendorff preparation).71 Our results from the in vivo experiment showed that there were significant differences in the mean heart rate of the two groups (control animals: 307±9 bpm; animals with epilepsy: 346±7 bpm). In contrast, we did not find these differences in the isolated ex vivo experiment (control animals: 175±7 bpm; animals with epilepsy: 176±6 bpm). These findings suggest that some part of the central nervous system (e.g., the thalamic nuclei) has the potential to modulate cardiovascular function, which could explain the pathophysiology underlying SUDEP. In a more recent study, our group (Scorza and colleagues, unpublished data) evaluated the heart rate (in vivo and isolated ex vivo) of rats with epilepsy before and after bilateral lesioning of the lateral posterior nuclei of the thalamus. The results showed significant differences in the mean heart rate before and after thalamic injury in the in vivo experiment. Surprisingly, no differences in heart rate could be observed before and after thalamic injury in the isolated ex vivo experiment. These observations seem to indicate the existence of a specific thalamic modulation of cardiac functioning that could support our hypothesis that SUDEP occurs due to cardiovascular dysfunction secondary to thalamic dysfunction. Finally, based on these results, we raised the possibility that the presence of thalamic nuclear lesions in people with temporal lobe epilepsy could be responsible for some of the processes that culminate in SUDEP, and that cardiovascular dysfunction could play a significant role in this condition.72
Taking all of these data together, the important question arises as to how to use our growing understanding of thalamic abnormalities and cardiovascular dysfunction in epilepsy patients to guide our examination of the possible role that abnormalities in thalamic nuclei play in the development of cardiovascular abnormalities in schizophrenic patients73. However, from post-mortem studies, it is unclear whether thalamic abnormalities or antipsychotic treatment contribute to sudden cardiac death, since most of the patients in these studies are treated with typical and atypical antipsychotics for several decades.
In schizophrenic patients, treatment with antipsychotics not only induces QT interval prolongation, but also influences thalamic volume and activity. The first study on this topic found that both typical and atypical antipsychotic treatment increased thalamic volumes in schizophrenic patients.20 A recent MRI study also found that the use of atypical antipsychotics is associated with an enlargement of the thalamus.74 However, in another study, the authors found that after patients switched from typical neuroleptics to the atypical antipsychotic olanzapine, their thalamic volume decreased to a normal size as compared to healthy controls.75 Another study found that patients who were taking atypical antipsychotics had decreased thalamic volumes, whereas patients treated with typical neuroleptics did not.76 The atypical antipsychotic risperidone has been shown to increase levels of the neuronal marker NAA in the thalamus.77 Accordingly, PET studies have shown an increased regional cerebral blood flow in the thalamus after treatment with the typical neuroleptic haloperidol, whereas olanzapine reduced blood flow in this region.78 In animal studies, the typical antipsychotic haloperidol as well as the atypical drugs clozapine and olanzapine have been shown to induce c-fos expression in the thalamus, which is related to activation of gene expression in cells in which its expression is induced.79
Hyperactivity of the dopamine D2 neurotransmitter system has also been implicated in schizophrenia. This increased dopaminergic signaling may result in disinhibition of the thalamus.80 In a PET study, haloperidol was found to downregulate the activity of DOPA decarboxylase in the thalami of schizophrenic patients, suggesting that haloperidol leads to a decrease in the dopamine synthesis capacity of the thalamus.81 Additionally, the occurrence of akathisia (as a side effect of olanzapine treatment) reduced the level of metabolic activity that was present in the thalamus of affected patients.82 MRI spectroscopy studies have indicated that high glutamine levels in the thalamus are reduced after atypical antipsychotic treatment is initiated.56,57 Animal models of antipsychotic treatment have demonstrated an activation of inhibitory GABAergic neurons, a decreased GABAA receptor binding, and an altered expression of proteins that interact with glutamate transporters.83–85 In summary, the inhibition of thalamic activity may be a mode of action of at least atypical antipsychotics and may influence patients’ sensitivity to cardiac events.
Where do we go from here? A priority of ours is to raise the possibility that the presence of lesions in the thalamic nuclei of patients with schizophrenia could represent the underlying mechanism of some processes that culminate in sudden cardiac death in these patients. We also feel it is important to raise the possibility that heart failure may play a significant role in this mechanism. Antipsychotic treatment may contribute to bradyarrhythmias in schizophrenic patients. It is clear that more animal model and clinical research needs to be done on this topic. However, in a similar manner to what our group has reported about the relationship between heart issues and epilepsy, we believe that a clear relationship exists between schizophrenia, thalamic dysfunction, heart failure, and sudden cardiac death.
The authors would like to thank FAPESP, CInAPCe-FAPESP, CNPq, and FAEPA for supporting this study.