






Gastro-entero-pancreatic (GEP-NET) and thoracic neuroendocrine tumours (NETs) are one of the most heritable groups of neoplasms in the body, being multiple endocrine neoplasia syndrome type 1 (MEN1), the genetic syndrome most frequently associated with this type of tumours. Moreover, Von Hippel Lindau syndrome, tuberous sclerosis, type 4 multiple neoplasia syndrome, and type 1 neurofibromatosis are associated with an increased risk of developing GEP-NETs. Another important aspect in GEP-NETs and thoracic NETs is the knowledge of the molecular background since the molecular profile of these tumours may have implications in the prognosis and in the response to specific treatments. This review summarizes the main indications for performing a genetic study in patients with GEP-NETs and thoracic NETs, and the methods used to carry it out. Moreover, it offers a description of the main hereditary syndromes associated with these NETs and their molecular background, as well as the clinical implications of the molecular profile.
Los tumores neuroendocrinos (TNE) gastro-entero-pancreáticos (TNE-GEP) y torácicos son uno de los grupos de neoplasias más heredables del organismo, siendo el síndrome de neoplasia endocrina múltiple tipo 1 el síndrome genético que más frecuentemente se asocia a este tipo de tumores. Por otra parte, el síndrome de Von Hippel Lindau, la esclerosis tuberosa, el síndrome de neoplasia múltiple tipo 4 y la neurofibromatosis tipo 1 también están asociados a un mayor riesgo de desarrollar TNE-GEP. Otro aspecto importante en los TNE-GEP y TNE torácicos es el conocimiento del perfil molecular, ya que el perfil molecular de estos tumores puede tener implicaciones en el pronóstico y en la respuesta a tratamientos específicos. En esta revisión se resumen las principales indicaciones de solicitar estudio genético en pacientes con TNE-GEP y TNE torácicos, y los métodos empleados para su realización. Además, se ofrece una descripción de los principales síndromes hereditarios asociados a estos TNE y de su perfil molecular, así como de las implicaciones clínicas del perfil molecular.
Neuroendocrine tumours (NET) are a diverse and heterogeneous set of cancers that share a common origin, from cells in the diffuse neuroendocrine system, although they may differ markedly in many features, including their location, secretory activity, degree of differentiation and presence or absence of metastases.1 The most prominent group of NET are gastro-entero-pancreatic NET (GEP-NET), which in turn include gastrointestinal NET and pancreatic NET (PNET), pulmonary NET, thymus NET, thyroid NET, other NET of unknown origin, paragangliomas and pheochromocytomas. NET are one of the most heritable groups of neoplasms, occurring in at least ten genetic syndromes, with the most commonly associated hereditary syndrome being multiple endocrine neoplasia syndrome type 1 (MEN1).2,3
A combination of the advances in classic techniques such as immunohistochemistry, clinical biochemistry and imaging tests, and the study of the molecular biology of NET has provided us with a deeper and more precise understanding of these tumours. Mutations and specific alterations in genes that underlie the genesis of NET have been identified through advanced genetic analyses. Knowing the genetic background of NET has implications, not only in terms of genetic counselling, but also in predicting the response to specific treatments and in personalising our patients' treatment and follow-up.2,4–6 Due to the advances in molecular medicine techniques in recent years, a wide range of previously unknown germline genetic mutations involved in the development of some NET have been discovered, with potential clinical implications.7,8 In addition, the availability of next-generation sequencing (NGS) techniques has made it possible to analyse multiple genes quickly and simultaneously, which can be very useful in patients with NET, who may present in the context of multiple hereditary predisposition syndromes.9
This paper summarises the main indications for requesting a genetic study in patients with GEP-NET and thoracic NET. A description of the main inherited genetic syndromes associated with these NET and their molecular profile is also provided, including the clinical implications of the molecular profile in GEP-NET and thoracic NET. The main strategies used for the genetic study in patients with hereditary syndromes (targeted tests or gene panels and NGS techniques) are also described, and the advantages and disadvantages in each case are briefly pointed out.
GEP-NETMolecular profileGEP-NET are the largest group of NET, accounting for 50% of all NET. An estimated 5%–10% of cases occur in the context of hereditary syndromes associated with NET.10 There is a relatively large amount of molecular and genetic information available for PNET and intestinal NET, although it is limited in the case of gastric NET.
The distinction between NET and neuroendocrine carcinoma (NEC) is linked to their genetic origin. The key difference between NEC and NET is the inactivation of the p53 and RB1 proteins.11 The presence of a mutation in p53 or loss of RB1 expression in the histomorphological analysis is useful in differentiating grade 3 PNET from NEC.12
In the case of sporadic PNET, recurrent somatic mutations have been found in the MEN1 tumour suppressor gene in up to 40% of patients,13 in chromatin remodelling genes such as ATRX/DAXX in 38%6 and in the PTEN/TSC 1-2 /DEPDC5 complex in 15%. The mutational profile also seems to be different between functioning and non-functioning PNET, as the prevalence of MEN1 mutations in glucagonomas is 60%, 40% in gastrinomas and 2%–20% in insulinomas.14 However, in sporadic intestinal NET, mainly chromosomal abnormalities are identified, with somatic mutations in CDKN1B and APC being identified in a minority of cases (approximately 10%).15 Somatic mutations in CDKN1B are one of the most specific gene mutations for intestinal NET. CDKN1B codes for a cyclin-dependent kinase inhibitor which binds to and inhibits Cdk2 and Cdk4.16,17 The APC/β-catenin pathway has been implicated in the initial progression of multipotent stem cells to neoplastic cells in these tumours.18
Additionally, epigenetic abnormalities play an important role in the biology of GEP-NET. Several genes are differentially methylated in significant subsets of GEP-NET, and overall DNA hypomethylation is a mechanism proposed as a driver of chromosomal instability in association with ALT activation in PNET.19 In intestinal NET, DNA methylation studies have found three subgroups with different methylation patterns with different prognosis.20
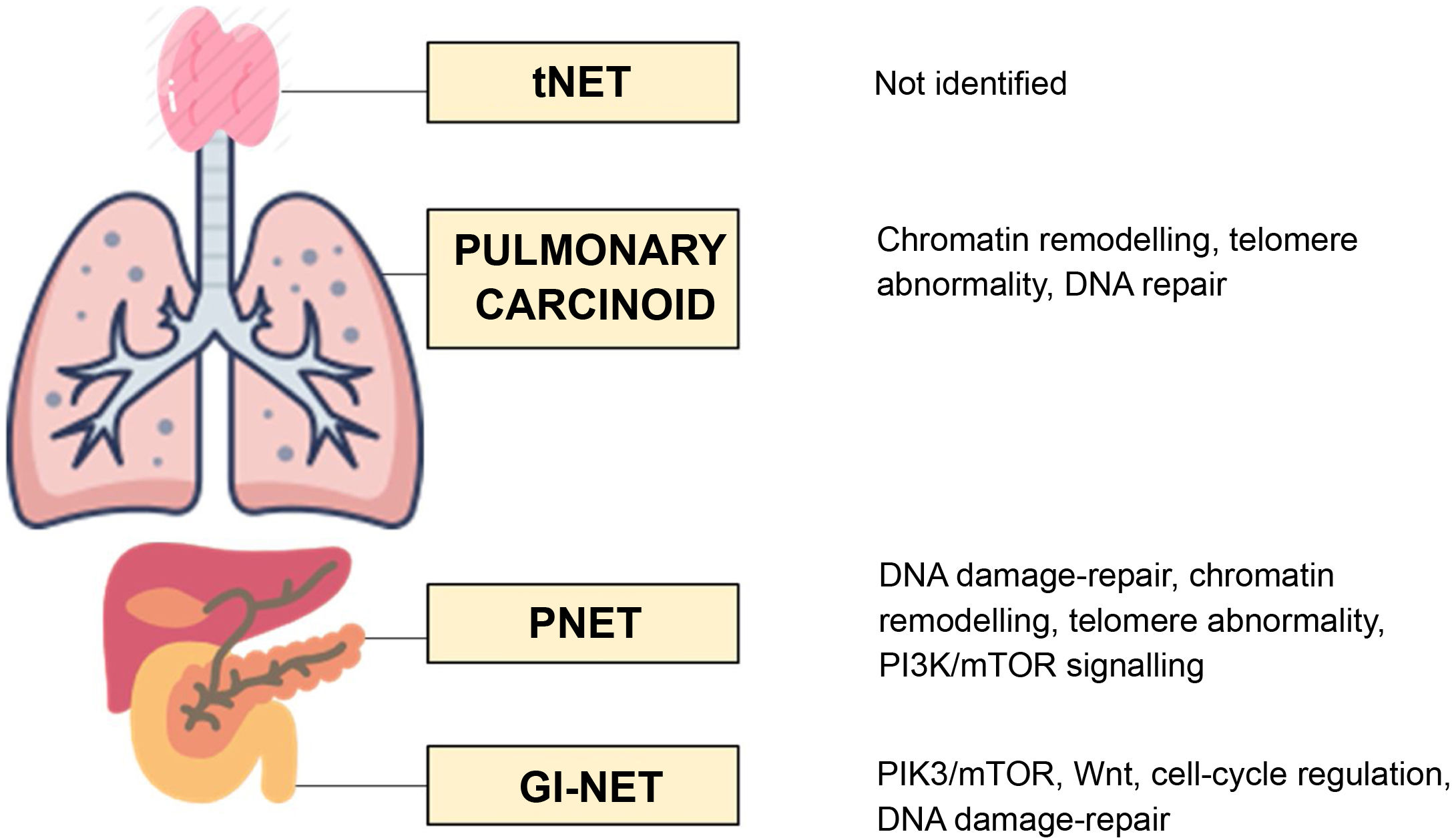
The three molecular pathways primarily involved in the development of GEP-NET are the DNA damage-repair pathway, the cyclin-dependent cell-cycle regulation pathway and the PI3K-AKT-mTOR pathway. Up to five altered pathways have been identified in PNET: the DNA damage-repair pathway; the cell-cycle regulation pathway; the chromatin-remodelling pathway; the telomere pathway; and the PI3K-AKT-mTOR-signalling pathway. In contrast, in small intestine NET, only the PIK3-AKT-mTOR pathway, the Wnt pathway, the cell-cycle regulation pathway and the DNA damage-repair pathway are involved15 (Fig. 1).
Clinical implications of the molecular study in GEP-NET
Molecular markers associated with a good prognosis have been identified, such as the expression of endothelial growth factor or somatostatin receptor type 2; with a better response to chemotherapy in PNET with loss of expression of retinoblastoma, and others associated with worse prognosis such as mutations in ATRXX/PDX121 or loss of p16 immunostaining. Some of these molecular abnormalities may also be specific markers of response to targeted therapy, such as alterations in the AKT-mTOR pathway, which have been associated with a better response to temsirolimus.22 Therefore, knowledge of these molecular markers could be useful for personalising follow-up and guiding the selection of systemic treatments.23
Two tools have been created based on the molecular profile of NET with promising utility for diagnosis, prognosis and response prediction in NET: the NETest and the PPQ. The role of the NETest has been widely studied. This study is performed on peripheral blood from which messenger RNA is extracted and subsequently transformed into DNA to measure gene expression by PCR. A score of 0–100 is given according to gene expression. The results obtained in the NETest are useful for the precise diagnosis of NET, the real-time follow-up of the disease status, the prediction of aggressive tumour behaviour, identification of the degree of surgical resection of the tumour and for the prediction of response to medical and radionuclide therapy in GEP-NET, pulmonary NET, pheochromocytomas and paragangliomas.24 The PPQ is a molecular liquid biopsy based on 8 growth factors and NET-specific genes which makes it possible to predict the efficacy of radionuclide therapy with 95% accuracy.25
Genetic syndromes associated with GEP-NETAlthough GEP-NET are of genetic origin in less than 10% of patients, forming part of typical hereditary syndromes such as MEN1 syndrome or multiple endocrine neoplasia type 4 (MEN4), von Hippel Lindau syndrome (VHL), tuberous sclerosis (TS) or neurofibromatosis type 1 (NF1) (Table 1), germline mutations in MEN1, VHL and CDKN1B have been described in 17% of apparently sporadic PNET. Germline mutations of DNA damage-repair genes, such as MUTYH, CHEK2, and BRCA2, have also been detected at a rate of approximately 6% in GEP-NET.5 Other germline mutations which have been described in PNET include APC, RAD50, RECQL4, FANCC, MAPKBP1, and PIF1 in <5%.7,8 However, many of these mutations have low penetrance or uncertain clinical activity, such as RAD50.8
Hereditary syndromes associated with GEP-NET.
| Genetic syndrome | Gene | Chromosome | Protein | Inheritance | Penetrance |
|---|---|---|---|---|---|
| MEN1 | MEN1 | 11q13.1 | Menin | ADI | >95% |
| 10% de novo mutations | |||||
| MEN4 | CDKN1B | 12p13.1 | P27 | ADI | Unknown |
| VHL | VHL | 3p25.3 | VHL | ADI | 50% at age 50 |
| 20% de novo mutations | |||||
| TS | TSC | TSC1:9q34.13 | Hamartin | ADI | 100% |
| TSC2: 16p13.3 | Tuberin | 75% de novo mutations | |||
| NF1 | NF1 | 17q11.2 | Neurofibromina | ADI | 100% (childhood) |
TS: tuberous sclerosis; ADI: autosomal dominant inheritance; MEN1: multiple endocrine neoplasia syndrome type 1; MEN4: multiple endocrine neoplasia syndrome type 4; NF1: neurofibromatosis type 1; GEP-NET: gastro-entero-pancreatic neuroendocrine tumour; VHL: von Hippel Lindau syndrome.
MEN1 is an autosomal dominant inheritance (ADI) syndrome caused by germline mutations on chromosome 11q13, whose typical triad is the association of primary hyperparathyroidism (PHPT) caused by multiglandular disease (95%), pituitary adenomas (20%–40%) and pancreatic or duodenal NET (40%–80%).26De novo mutations occur in up to 10% of cases, with no family history of MEN1.27 However, among patients who meet the clinical criteria for MEN1, approximately 10% have no identifiable mutations.27
PNET are the second most common tumour in MEN1 and the main cause of death in these patients, with an estimated survival rate 10 years after diagnosis ranging from 23% to 62%.28 The lifetime risk of developing a PNET in patients with MEN1 is estimated to be 40%–80%.28 The most common PNET in MEN1 are nonfunctioning. Nonfunctioning PNET are usually multifocal; diagnosis is made at an earlier age than their sporadic counterparts (mean age at diagnosis 40) and they have a more indolent disease course than sporadic cases. They are usually asymptomatic when diagnosed but they can metastasise, and the liver is the most common metastasis site.29 A two-fold higher risk of developing PNET and a significantly higher frequency of metastatic disease has been found in patients with mutation in exon 2 than in patients without mutation in exon 2 (53% vs 23%, P = .049).30
Among the functioning GEP-NET in MEN1, the most common one by far is gastrinoma. In fact, an estimated 25%–30% of gastrinomas are associated with MEN1, and up to 60% of patients with MEN1 will develop a gastrinoma during their lifetime.31,32 In patients with MEN1, gastrinomas occur an average a decade earlier than in sporadic cases,1 they are located almost exclusively in the duodenum and there is a higher incidence of severe oesophageal disease than in sporadic cases.33 Insulinomas are the second most frequent functioning GEP-NET in MEN1; approximately 15% of patients with MEN1 have insulinomas, which may be the first manifestation of the syndrome in up to 10%.34 Around 4%–10% of insulinomas are associated with MEN1. MEN1 insulinomas are usually smaller tumours than sporadic ones, but are more frequently metastatic (up to 25% of cases in MEN1 vs 10% in sporadic insulinomas).35 Other less common tumours such as glucagonomas, vipomas or somatostatinomas are detected in less than 5% of patients with MEN129 (Fig. 2).
MEN4 and GEP-NET
MEN4 is a variant of MEN1 also associated with GEP-NET and ADI. It shares a similar phenotype to MEN1 but is the result of mutations on chromosome 12p in CDKN1B, which codes for the p27 protein, which is a tumour suppressor gene that regulates cell-cycle progression.36 The tumours most commonly associated with MEN4 are pituitary and parathyroid. GEP-NET are less common in MEN4 than in MEN1 (occur in 25% of MEN4 versus 60%–80% in MEN1) and are usually non-functioning gastrinomas or PNET.37
VHL and GEP-NETVHL syndrome or disease is another of the hereditary syndromes associated with GEP-NET. It is an ADI syndrome caused by inactivating mutations in the VHL tumour suppressor gene, located on the chromosome 3p25.38 VHL disease shows marked phenotypic variability and age-dependent penetrance.39 It is characterised by a greater predisposition to haemangioblastomas of the central nervous system (CNS), paragangliomas, clear cell renal cell carcinoma, renal cysts, NET and primarily pancreatic cysts and tumours of the endolymphatic sac. This syndrome is subclassified according to the risk of pheochromocytoma into type 1 (no pheochromocytoma) and type 2 (high risk of pheochromocytoma). In turn, VHL type 2 includes three subtypes according to the risk of clear cell renal cell carcinoma: type 2A (without clear cell renal cell carcinoma), 2B (high risk of clear cell renal cell carcinoma) and 2C (only pheochromocytoma).38
With regard to the risk of PNET, although around 75% of patients with VHL develop some type of pancreatic lesion, the majority are cystic lesions, generally serous cystadenomas. PNET occur in 10%–17% of VHL,38 although the risk is higher in VHL patients with mutations in exon 3 of the VHL gene.40 However, overall, the risk of VHL in a patient with PNET is low, being approximately 0.5%. PNET in VHL are usually non-functioning, 30%–50% are multiple and they are generally diagnosed earlier than sporadic ones, which could partly explain the lower risk of malignancy described in patients with PNET in VHL than in patients with other sporadic PNET.41
Tuberous sclerosis and GEP-NETTuberous sclerosis (TS) is an autosomal dominant disorder characterised by multisystem manifestations, including hamartomas in the brain, heart, lung, kidneys and skin.42 Infrequently (in 1% of cases) it is associated with NET, generally pancreatic and non-functioning, and insulinomas.43 TS is caused by functional mutations in the tuberous sclerosis complex type 1 (TSC1) and type 2 (TSC2) genes, which encode proteins that form the tuberin-hamartin complex, essential for signalling the mTOR pathway (a pathway responsible for the regulation of cell growth, differentiation and proliferation).44 However, approximately 15%–20% of the patients who meet the clinical criteria for TS have no identifiable mutations.
Neurofibromatosis type 1 and GEP-NETNeurofibromatosis type 1 (NF1) or von Recklinghausen's disease is an ADI syndrome caused by mutations in the neurofibromatosis 1 gene located on the chromosome 17 and codes for a protein called neurofibromin. However, approximately 42% of affected individuals have de novo mutations.45
The clinical manifestations are mainly cutaneous and in the CNS, although around 1% also present GEP-NET. Most of them are located in the region of the ampulla of Vater in the duodenum and are generally somatostatinomas.46 However, they can be silent in a high percentage of cases and manifest with symptoms deriving from the tumour's mass effect. The coexistence of somatostatinoma and gastrointestinal stromal tumour in the same patient can be considered virtually pathognomonic for NF1. In PNET, insulinomas are the most common type. In general, they are less aggressive than the sporadic types in terms of the risk of metastasis.47
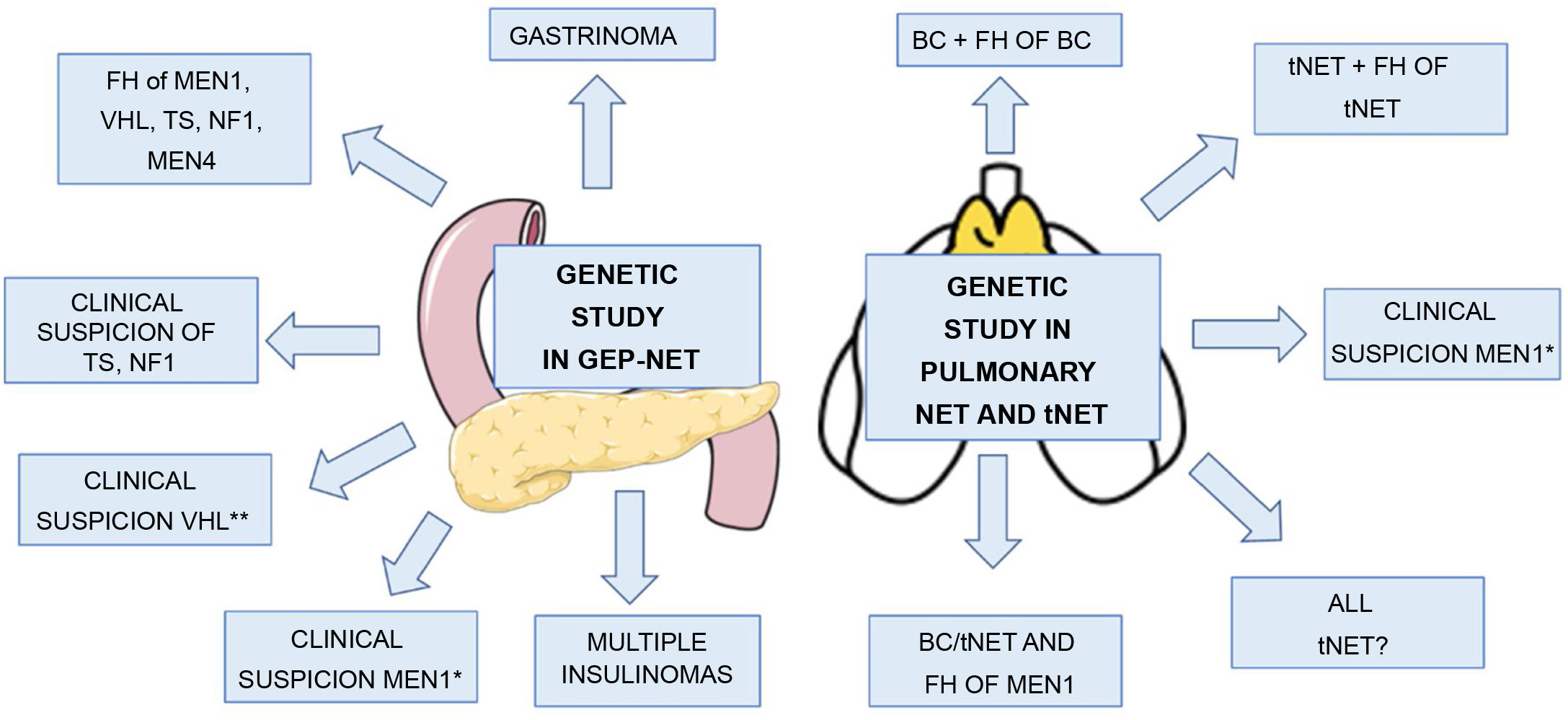
Indications for genetic study in GEP-NETThe indications for genetic study in patients presenting with GEP-NET include: 1) gastrinoma at any age, remembering that 25% of gastrinomas occur in patients with MEN131,32; 2) multiple insulinomas at any age (insulinomas in patients with MEN1 are generally small and numerous, sporadic ones are usually single); 3) clinical suspicion of MEN1, for example, coexistence of GEP-NET and PHPT or GEP-NET and pituitary adenoma or of two or more MEN1-related tumours26,48; 4) clinical suspicion of VHL, for example, GEP-NET, and one or more lesions associated with VHL (haemangioblastoma, clear cell renal cell carcinoma, pheochromocytoma, medial endolymphatic sac tumour, epididymal papillary cystadenoma, pancreatic serous cystadenoma)39,49; 5) GEP-NET in patients with clinical suspicion of NF1,45 TS42 or MEN4 (same criteria for MEN1 in patient with negative genetic study for MEN1); and 6) GEP-NET in patients with a family history of MEN1, VHL, NF1, TS or MEN410 (Table 2) (Fig. 3). A genetic study should also be considered in patients with GEP-NET who are diagnosed at a very young age (<30), as they could be the first manifestation of MEN1 or even of another of the hereditary predisposition syndromes previously described.
Diagnostic criteria of the main hereditary syndromes associated with GEP-NET.
| Hereditary syndromes | Clinical diagnosis |
|---|---|
| MEN126 | Clear criteria for MEN1: ≥2 of the 3 most common tumours (parathyroid, GEP-NET and pituitary) |
| No clear criteria for MEN1, but suspect/atypical MEN1: | |
| Two or more related tumours (parathyroid, enteropancreatic, pituitary, adrenal, lipomas, carcinoids, angiofibromas, collagenomas) | |
| Parathyroid tumours under age of 30 | |
| Recurrent primary hyperparathyroidism | |
| Gastrinoma at any age | |
| Multiple islet cell tumours at any age | |
| Isolated familial hyperparathyroidism | |
| MEN437 | Same criteria as MEN1 in patients with a negative genetic study for MEN1 |
| VHL38 | >2 VHL-associated lesions (haemangioblastoma, clear cell renal cell carcinoma, pheochromocytoma, medial endolymphatic sac tumour, epididymal papillary cystadenoma, pancreatic serous cystadenoma, PNET) |
| One lesion associated with VHL and a family history of VHL-associated lesions | |
| Haemangioblastoma diagnosed under age of 30 | |
| >2 haemangioblastomas diagnosed at any age | |
| Clear cell renal cell carcinoma diagnosed under age of 40 | |
| Bilateral or multiple clear cell renal cell carcinoma | |
| Pheocromocytoma under age of 40 | |
| Bilateral or multiple pheochromocytoma | |
| >1 pancreatic serous cystadenoma | |
| >1 PNET | |
| Multiple pancreatic cysts in any VHL-associated lesion | |
| Papillary epididymal cystadenoma at any age | |
| Bilateral epididymal cysts | |
| TS42 | Two or more major features: |
| Hypomelanotic macules (more than 2 and ≥5 mm in diameter) | |
| Angiofibromas (more than 2) or fibrous cephalic plaque | |
| Periungual fibromas (more than 1) | |
| Shagreen patch | |
| Multiple retinal hamartomas | |
| Cortical dysplasias | |
| Subependymal nodules | |
| Subependymal giant cell astrocytoma | |
| Cardiac rhabdomyoma | |
| Lymphangioleiomyomatosis | |
| Angiomyolipomas (more than 1) | |
| One major feature with at least 2 minor features: | |
| Confetti-like skin lesions | |
| Tooth enamel pitting (more than 3) | |
| Oral fibromas (more than 1) | |
| Retinal achromic patch | |
| Multiple renal cysts | |
| Non-renal hamartomas | |
| NF145 | 2 or more major criteria required for diagnosis: |
| Six or more café-au-lait spots (>0.5 cm in children or >1.5 cm in adults) | |
| Two or more cutaneous or subcutaneous neurofibromas or one plexiform neurofibroma | |
| Axillary or inguinal freckling | |
| Optic pathway glioma | |
| Two or more Lisch nodules | |
| Bone dysplasia | |
| First-degree relative with NF1 |
TS: tuberous sclerosis; MEN4: multiple endocrine neoplasia syndrome type 4; MEN1: multiple endocrine neoplasia syndrome type 1; NF1: neurofibromatosis type 1; PNET: pancreatic neuroendocrine tumour; GEP-NET: gastro-entero-pancreatic neuroendocrine tumour; VHL: von Hippel Lindau syndrome.

Indications for genetic study in GEP-NET and pulmonary NET and tNET.
FH: family history; BC: bronchial carcinoid; TS: tuberous sclerosis; NF1: neurofibromatosis type 1; VHL: Von Hippel Lindau; NET: neuroendocrine tumour.
*Coexistence of GEP-NET and PHPT or GEP-NET and pituitary adenoma or two or more MEN1-related tumours.
** Haemangioblastoma, clear cell renal cell carcinoma, pheochromocytoma, medial endolymphatic sac tumour, epididymal papillary cystadenoma, pancreatic serous cystadenoma.
As far as pulmonary NET are concerned, bronchial carcinoids (BC) account for 2% of primary lung neoplasms. Neuroendocrine lung carcinomas and BC have a differential molecular profile, with mutations in p53 and RB1 being the most useful markers indicative of NEC.50 In the case of BC, recurrent mutations in MEN1 have been identified in up to 40% of patients, and loss of heterogeneity and mutations in PIK3CA in 13%–40%.50 The main pathways involved in the pathogenesis of BC are the chromatin-remodelling, DNA damage-repair and telomere pathways (Fig. 1).
With regard to the patterns of chromatin remodelling and DNA methylation, few studies have focused on this aspect in pulmonary NET, although a negative association has been described between the nuclear overexpression of the arginine methyltransferase-5 protein (PRMT5) and tumour grade,51 and the presence of a differential methylation pattern between typical and atypical BC and pulmonary neuroendocrine carcinomas.52
Clinical implications of the molecular study in pulmonary NETCertain molecular and genetic alterations in BC have been associated with survival outcomes and others could be potential markers of response to specific therapies. Knowledge of the molecular profile in BC is of great importance, both for the stratification of the risk of progression and for the personalisation of targeted treatment in cases that are not candidates for surgery or with tumour persistence or progression.53 For example, mutations in MEN154 are associated with a poor prognosis, regardless of gender, age and being a smoker or non-smoker; mutations in ATRX have been associated with poorer survival (hazard ratio = 11, 95% CI, 1.8–68, P = .01)55; chromosomal instability is a common event in atypical and metastatic carcinoids, and mutations in PI3K-AKT-mTOR are associated with greater aggressiveness in BC.56 Mutations in the PIK3 pathway may also be a potential marker of response to everolimus.57,58
Genetic syndromes associated with BCUp to 5% of BC present in the context of MEN1 and are much less frequent in the context of familial pulmonary carcinoid syndrome.51
MEN1 and BCMEN1 can involve BC in up to 13% of patients, while around 5% of BC occur in patients with MEN1.59 However, the prevalence of BC in MEN1 depends on the criteria used for the diagnosis of BC. The prevalence is 6.7% when only cases of BC verified by histology are included, and around 30% when small bronchopulmonary nodules observed on CT are also included.60
Compared to sporadic BC, those associated with MEN1 are diagnosed earlier (age 42-40 vs age 64),61 which may partly explain why they are associated with lower mortality rates. There do not seem to be gender-related differences in prevalence in MEN1. They are usually asymptomatic and tend to be discovered in the screening imaging tests routinely requested in the investigation of MEN1. They generally follow an indolent course, as most of these tumours are well-differentiated NET (typical and atypical BC). BC do not therefore reduce the overall survival of patients with MEN1.62 As in BC not associated with MEN, survival is better in females than in males, in typical rather than atypical BC, and in BC without metastasis rather than metastatic BC.63
Familial bronchial carcinoid syndromeThe other genetic syndrome, much less known, is familial BC syndrome, which has only been reported in one study published in 2001 describing two families without mutations in MEN1, in which two cases of BC in patients between the ages of 70 and 80 were found in each family, without any other manifestations of MEN1, with the existence of another type of as yet unknown genetic susceptibility being suspected.64
Indications for genetic study in BCThe indications for requesting a genetic study in BC would be: 1) pulmonary NET with a family history of MEN1; 2) pulmonary NET with a family history of BC; 3) patients with pulmonary NET and clinical suspicion of MEN1; and 4) pulmonary NET in a patient with other tumours associated with MEN1 (coexistence of GEP-NET and PHPT or GEP-NET and pituitary adenoma or two or more MEN1-related tumours)26 (Table 2). Most of the pulmonary NET in MEN are BC, so the genetic study should be considered mainly in patients with BC and not in other types of pulmonary NET unless there is a family history of MEN1.
NET of the thymus (tNET)Molecular profile and clinical implications of the molecular study in tNETtNET account for 0.5% of all NET. The study by Sakane et al.65 reports that the most frequently mutated genes in tNET are p53 (18.5%), followed by KIT (7.4%) and PDGFRA (5.6%). In terms of the molecular profile, no potential prognostic biomarkers or targetable molecular abnormalities in these tumours have been identified as yet.65,66 Understanding of the methylation profile in tNET is very limited. One study focusing on this aspect in tNET found that RASSF1A was strongly hypermethylated in NET, but not in thymic carcinomas or thymomas. They also found that low-grade NET tumour tissue was more strongly methylated than high-grade.67
Familial syndromes associated with tNETMEN1 and tNETThe only hereditary syndrome associated with an increased risk of tNET is MEN1. In fact, around 25% of tNET occur in the context of MEN1 and 3.7% (range 2.0%–8.2%) of MEN1 develop a tNET during follow-up.68 More than half of tNET have metastatic disease at diagnosis.68 It is one of the leading causes of death in MEN1; a recent study reported that 19% of deaths in MEN1 were due to tNET.69 Survival at five years is 69.5%.
Several factors associated with a worse prognosis have been identified, such as older age (over 43.0 years), a larger maximum tumour diameter (>5.0 cm) and the presence of metastases.68 For all these reasons, it is important to follow the recommendations for screening for bronchial and thymic NET, and perform a chest CT or MRI every 1–2 years, as early diagnosis is associated with longer survival.
Indications for genetic study in tNETThe indications for a genetic study are similar to those of BC: 1) patients with a family history of MEN1 or tNET; and 2) clinical suspicion of MEN126 (Table 2). However, considering that 25% of tNET occur in the context of MEN, that they are one of the main causes of death in MEN1, and that early diagnosis is associated with longer survival, requesting genetic screening for MEN1 should be considered in all patients with tNET.
Strategies for carrying out the genetic study in hereditary syndromesAlthough the current guidelines generally recommend the use of targeted testing or small-scale gene panels for the identification of hereditary endocrine tumour genetic susceptibility syndromes, NGS techniques which are widely available in routine clinical practice have been developed. The application of these techniques makes it possible to analyse multiple genes quickly. The improvement in the diagnostic accuracy of current molecular techniques has enabled a significant proportion of patients with apparently sporadic NET to be classified as hereditary NET. Therefore, in patients with high clinical suspicion of hereditary syndromes with a negative genetic study carried out years ago, consideration should be given to repeating the genetic screening if more advanced techniques are available and previously unknown genes should be included (for example, CDKN1B). The genetic study has implications for diagnosis and tumour risk assessment with a view to the selection of therapy, follow-up and planning and screening of relatives. Considering that early diagnosis of most GEP and thoracic NET is associated with increased survival, genetic testing of asymptomatic family members is recommended in order to establish an early diagnosis.70
Targeted genetic testing/gene panelFor targeted genetic testing, conventional Sanger sequencing is the most widely used technique and is considered the gold standard.71 The Sanger technique is considered relatively quick and cheap if the genomic region of interest is small, but it becomes a low-throughput technique if the genes under study are long or numerous, as in this case it is labour-intensive, time-consuming and expensive. However, this is the recommended technique in cases of family screening in asymptomatic relatives with a family history of an already identified genetic abnormality or in patients with symptoms indicative of a specific genetic syndrome.70
Next-generation sequencing techniquesNGS is capable of investigating more than 50 genes simultaneously, often at a lower cost than targeted testing.9 The main drawbacks of this technique are that not all genes included in gene panels are always of significant clinical relevance and certain findings can be identified in genes with no clearly established clinical value that can be difficult to interpret. Interpretation criteria for sequence variants as “pathogenic”, “probably pathogenic”, “variant of uncertain significance”, “probably benign” and “benign” have been defined by the American College of Medical Genetics and Genomics in a joint consensus recommendation.72 NGS should be considered as a first-line technique in patients in whom a tumour may be part of several hereditary syndromes (for example, pheochromocytomas or paragangliomas).70 Therefore, taking into account the indications for genetic study in GEP-NET, pulmonary NET and tNET, this technique will be used in a minority of cases.
ConclusionsThe molecular profile of GEP-NET and thoracic NET has prognostic implications and predicts response to targeted therapy. Advances in molecular medicine techniques, with the discovery of new genes and a better understanding of the ones we already know, are opening up the field to increasingly personalised precision medicine, allowing both the prognosis of patients with a NET to be predicted based on molecular profile as well as the options of response to specific treatments.
The hereditary syndromes typically associated with GEP-NET are MEN1, MEN4, VHL, TS and NF1. About 5% of pulmonary NET and 25% of tNET occur in the context of MEN1. It is important to be familiar with the criteria for carrying out a genetic study in these tumours in order to offer the patient adequate genetic counselling and personalised follow-up.
FundingThis study did not receive any type of funding.
Conflicts of interestThe author declares that she has no conflicts of interest.
